When the adrenal cortex acts as the hypothalamus: a rare case of ectopic corticotropin-releasing hormone production

 CC BY 4.0
CC BY 4.0
When the adrenal cortex acts as the hypothalamus: a rare case of ectopic corticotropin-releasing hormone production
A 73‑year‑old woman was admitted to a cardiology department with a suspicion of non–ST‑segment elevation myocardial infarction (NSTEMI). She complained of progressively worsening leg edema and dyspnea that lasted for a month. Her medical history included hypertension, hyperlipidemia, meningioma, lung nodules, and hepatitis C infection. She was receiving a β-blocker, statin, and antihypertensive drugs. While NSTEMI was ruled out, the patient was referred to an endocrinology unit due to suspected hypercortisolemia. Physical examination showed abdominal obesity, a buffalo hump, and easy bruising. Biochemical test results were consistent with ectopic adrenocorticotropic hormone / corticotropin‑releasing hormone (ACTH/CRH) secretion (Supplementary material, Table S1); however, the findings of extensive imaging studies were inconclusive (Figure 1A). Neither the computed tomography scan of the abdomen, pelvis, and thorax nor magnetic resonance imaging of the head and pituitary gland revealed potential source of ectopic ACTH/CRH secretion.
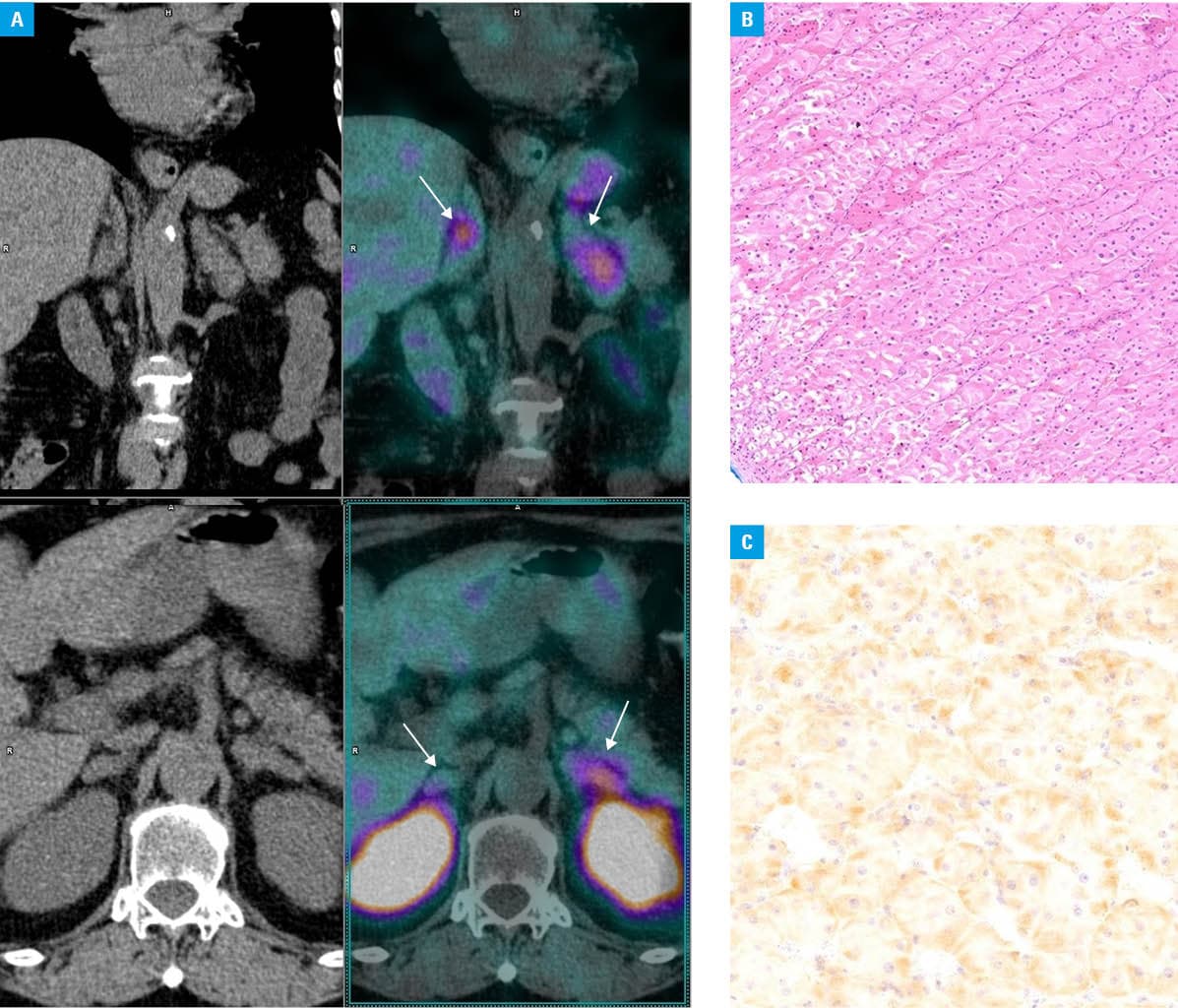
The patient was diagnosed with diabetes mellitus and osteoporosis. Treatment included intensive insulin therapy, antiresorptive medications, and correction of hypokalemia. She developed severe pneumonia that was treated with antibiotics. Metyrapone therapy was initiated, and the patient required rapid dose escalation, up to a maximum of 2500 mg daily. Given the diagnosis of severe Cushing syndrome (CS) with limited response to medical therapy, bilateral adrenalectomy was performed.
Histological examination revealed diffuse adrenal cortex hyperplasia, with immunohistochemical staining negative for ACTH but positive for CRH (Figure 1B and 1C). Hydrocortisone replacement therapy was initiated. Treatment resulted in improved glycemic control and correction of electrolyte imbalance. Finally, insulin therapy was discontinued, and antihypertensive medications were reduced in both dosage and number. Following adrenalectomy, ACTH levels normalized and remained stable during 5‑year follow‑up (Supplementary material, Figure S1).
Ectopic CS (ECS) is an exceptionally rare disorder characterized by excessive hypercortisolemia.1 The clinical presentation varies depending on the histological type and aggressiveness of the primary tumor.2
In the described patient, bilateral adrenal hyperplasia with CRH expression was identified following adrenalectomy, which could have served as a potential cause of ECS. However, increasing evidence suggests the presence of intra‑adrenal CRH circuits. Moreover, chromaffin cells and pheochromocytomas may express CRH‑loop components. In certain instances, blood levels of these peptides may be sufficiently high to exert systemic effects, resulting in ECS development.3 Consequently, in this case, intra‑adrenal CRH secretion might have been significant enough to provoke hypercortisolism.
Alternatively, the patient’s condition could be attributed to an occult primary tumor that led to cyclic CS, which is characterized by alternating phases of elevated cortisol levels (peaks) followed by spontaneous phases of normal or reduced cortisol production (troughs). Cyclicity is defined as experiencing at least 1 trough occurring between 2 peaks, with the duration of these phases ranging from days to years—the longest recorded trough lasted 5 years.4 Despite similarity in symptoms and comorbidities between cyclic and noncyclic CS, the former is most commonly caused by pituitary tumors.4
Contrary to typical presentations, our patient has shown no clear signs of cyclicity during nearly 5 years of follow‑up. However, elevated serum ACTH concentrations might be the only indication of cyclicity in this case.
CS is associated with serious complications and significant morbidity.5 Identifying the tumor responsible for ECS development remains a significant challenge.
- Young J, Haissaguerre M, Viera‑Pinto O, et al. Management of endocrine disease: Cushing’s syndrome due to ectopic ACTH secretion: an expert operational opinion. Eur J Endocrinol. 2020; 182: R29‑R58. | Crossref
- Hayes AR, Grossman AB. The ectopic adrenocorticotropic hormone syndrome: rarely easy, always challenging. Endocrinol Metab Clin North Am. 2018; 47: 409‑425. | Crossref
- Tsatsanis C, Dermitzaki E, Venihaki M, et al. The corticotropin‑releasing factor (CRF) family of peptides as local modulators of adrenal function. Cell Mol Life Sci. 2007; 64: 1638‑1655. | Crossref
- Nowak E, Vogel F, Albani A, et al. Diagnostic challenges in cyclic Cushing’s syndrome: a systematic review. Lancet Diabetes Endocrinol. 2023; 11: 593‑606. | Crossref
- Valassi E, Tabarin A, Brue T, et al. High mortality within 90 days of diagnosis in patients with Cushing’s syndrome: results from the ERCUSYN registry. Eur J Endocrinol. 2019; 181: 461‑472. | Crossref
SUPPLEMENTARY MATERIAL
ARTICLE INFORMATION