Peutz–Jeghers syndrome (PJS) is an autosomal dominant genetic disorder caused by a mutation in the STK11 gene on chromosome 19.1 It is characterized by the development of multiple hamartomatous polyps in the gastrointestinal tract, which can lead to abdominal pain, chronic bleeding, and bowel obstruction.1 Other typical features of this syndrome include pigmentation of the mucous membranes and skin, as well as a predisposition to oncological diseases.1 PJS is a rare condition with an estimated prevalence ranging from 1/25 000 to 1/300 000 live births.2
A 72‑year‑old patient presented to a dermatology clinic due to the appearance of asymptomatic brown and dark blue to black pigmented mucosal macules on the upper and lower lips and oral mucosa of the cheeks, which had developed several months prior. The patient had a history of type 2 diabetes mellitus and arterial hypertension. Additionally, he had a pacemaker implanted due to second‑degree atrioventricular block. The patient reported no significant family history of PJS.
Three years ago, prostate biopsy was performed due to an elevated prostate‑specific antigen level (5.854 ng/ml; reference range, 0–4 ng/ml). Histopathologic examination confirmed the diagnosis of adenocarcinoma in the left lobe of the prostate. The Gleason score was 7, and the lesion constituted approximately 2% of the examined tissue. Scintigraphy showed no pathology. The cancer was staged as cT2cN0M0. The patient underwent transurethral resection of the prostate followed by radiotherapy. In the same year, a single polyp in the transverse colon was identified on colonoscopy, classified histologically as tubular adenoma with mild dysplasia. The lesion was completely removed.
One year later, the patient underwent another prostate biopsy due to recurrent hematuria. Meanwhile, numerous brown and dark blue‑black pigmented mucosal macules appeared on the upper and lower lips, spreading to the surrounding oral mucosa of the cheek (Figure 1A–1C). First, the hyperpigmented macules were present on the lower lip, and after a few weeks, they spread to the upper lip and buccal mucous membrane. Histopathologic examination of the prostate biopsy specimen demonstrated multiple fragments of the prostate gland with abundant chronic inflammation, some purulent glands, and adenocarcinoma in several sections. The Gleason score was 8, and the lesion constituted approximately 1% of the examined tissue. Leuprorelin (11.25 mg subcutaneously every 3 months) was initiated and continued until the dermatology appointment.
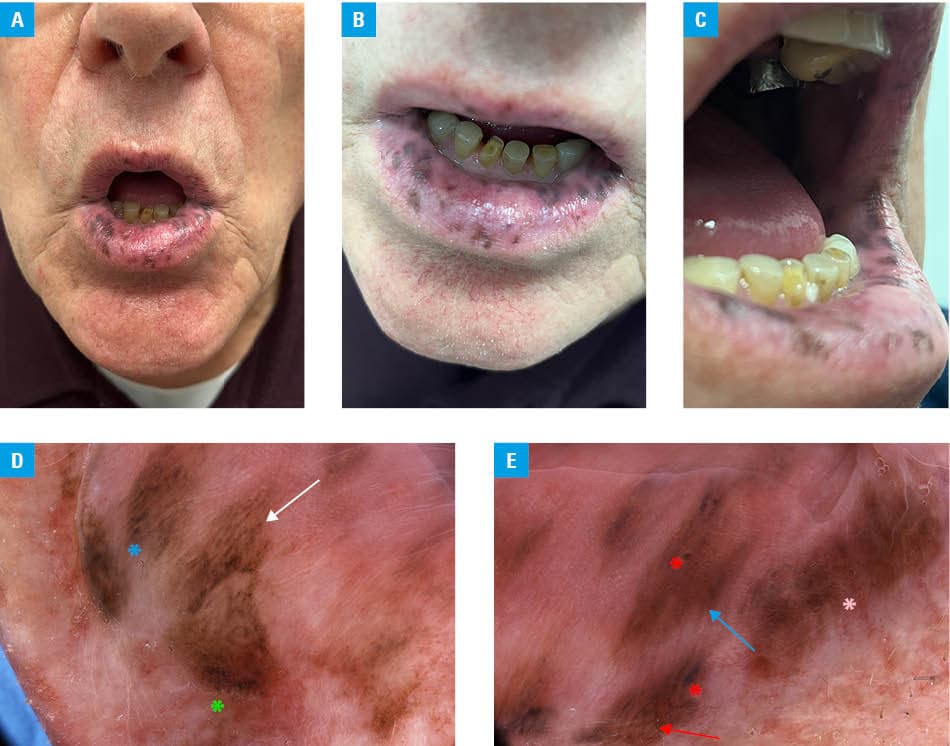
A clinical suspicion of PJS was raised by the dermatologist. Videodermoscopy identified brownish, black and grey dots, dark brown and black globules, as well as parallel, homogenous, and reticular patterns (Figure 1D and 1E). The patient continued treatment for prostate adenocarcinoma and remained under the care of urology and gastroenterology clinics.
PJS is associated with an increased risk of gastrointestinal cancers, including colorectal, small intestine, and pancreatic cancers.2,3 Extragastrointestinal cancers linked to this syndrome predominantly involve gynecological malignancies, such as ovarian, cervical, uterine, and breast cancers.2,3 In this case, the diagnosis of prostatic adenocarcinoma was notable, as prostate cancer is relatively uncommon in PJS.4,5 Moreover, the appearance of mucosal symptoms coincided with the recurrence of prostate cancer, being a clear sign of tumor progression. To the best of our knowledge, this is the first report presenting dermoscopy findings of pigmented mucosal lesions in this rare syndrome.
- Nevozinskaya Z, Korsunskaya I, Sakaniya L, et al. Peutz‑Jeghers syndrome in dermatology. Acta Dermatovenerol Alp Pannonica Adriat. 2019; 28: 135‑137. | Crossref
- Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz‑Jeghers syndrome. Clin Cancer Res. 2006; 12: 3209‑3215. | Crossref
- Chen HY, Jin XW, Li BR, et al. Cancer risk in patients with Peutz‑Jeghers syndrome: a retrospective cohort study of 336 cases. Tumour Biol. 2017; 39: 1010428317705131. | Crossref
- You YN, Wolff BG, Boardman LA, et al. Peutz‑Jeghers syndrome: a study of long‑term surgical morbidity and causes of mortality. Fam Cancer. 2010; 9: 609‑616. | Crossref
- Boardman LA, Thibodeau SN, Schaid DJ, et al. Increased risk for cancer in patients with the Peutz‑Jeghers syndrome. Ann Intern Med. 1998; 128: 896‑899. | Crossref
ARTICLE INFORMATION