Cutaneous vasculitis manifestations in patients with systemic lupus erythematosus: a comprehensive retrospective analysis with clinical implications
1,2 , 2,3, 2, 4, 5, 1,2
Key words: cutaneous vasculitis, skin vasculitis, systemic lupus erythematosus
, 2,3, 2, 4, 5, 1,2
Key words: cutaneous vasculitis, skin vasculitis, systemic lupus erythematosus

 CC BY 4.0
CC BY 4.0
Cutaneous vasculitis manifestations in patients with systemic lupus erythematosus: a comprehensive retrospective analysis with clinical implications
Introduction: Systemic lupus erythematosus (SLE) is an autoimmune disease with clinical and laboratory heterogeneity. Data on cutaneous vasculitis (CV) in SLE are limited, especially in the context of its clinical value.
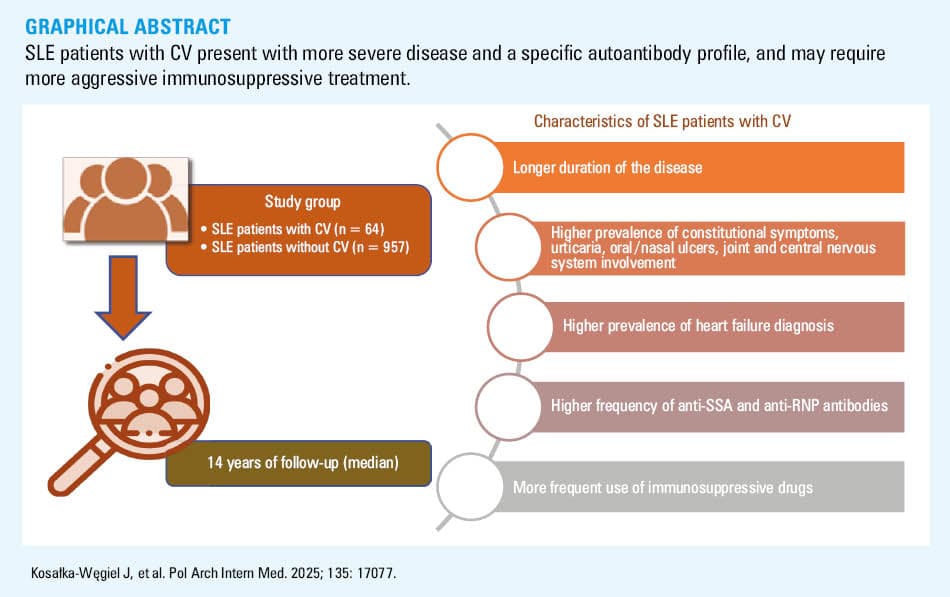
Objectives: This study aimed to compare the clinical characteristics, laboratory findings, and treatment patterns of SLE individuals with and without CV to determine if skin vasculitis identified a distinct subset of patients with unique outcomes.
Patients and methods: We conducted a retrospective analysis based on medical records of 1021 SLE patients (64 with CV and 957 without CV) treated at the University Hospital in Kraków, Poland, between 2012 and 2022. All patients met the 2019 European Alliance of Associations for Rheumatology / American College of Rheumatology classification criteria for SLE.
Results: Overall, CV was observed in 6.27% of the study cohort (n = 64). The patients with CV more often exhibited constitutional symptoms (87.5% vs 76.2%; P = 0.04), joint manifestations (96.9% vs 87.3%; P = 0.02), central nervous system (CNS) involvement (15.6% vs 6.6%; P = 0.007), and heart failure (14.1% vs 4.4%; P <0.001), as compared with the individuals without CV. Higher prevalence of anti–Sjögren syndrome type A (75% vs 59.2%; P = 0.02) and antiribonucleoprotein antibodies (35% vs 20.3%; P = 0.007) was observed in the CV group. Treatment involved more frequent use of azathioprine (51.6% vs 37.5%; P = 0.03), belimumab (9.4% vs 3.7%; P = 0.03), and cyclophosphamide (40.6% vs 27.5%; P = 0.02) in the individuals with CV, as compared with those without CV.
Conclusions: SLE patients with CV present with more severe disease, including heart failure and CNS involvement, and a specific autoantibody profile. These individuals may require more aggressive immunosuppressive treatment. Our findings suggest that CV in SLE may serve as a marker of more severe disease, necessitating careful monitoring and more intensive treatment.
What's new?
This study highlights the significance of cutaneous vasculitis (CV) as a marker of severe systemic lupus erythematosus (SLE). Our analysis of 1021 Polish patients with SLE (the largest cohort to date) showed that CV was associated with a longer disease duration, severe manifestations (such as heart failure and central nervous system involvement), and a specific autoantibody profile. Additionally, CV patients required more intensive immunosuppressive treatments, including azathioprine, belimumab, and cyclophosphamide. These findings underscore the need for early detection and aggressive management of CV in SLE to mitigate long‑term complications, especially damage to life‑critical organs, such as the heart and central nervous system.
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease affecting multiple organ systems. It is characterized by production of autoantibodies against nuclear antigens, deposition of immune complexes, and persistent inflammation targeting the skin, joints, and kidneys.1 The pathophysiology of SLE involves a complex interplay of genetic, environmental, and immune dysregulation factors, including defective clearance of immune complexes, impaired apoptotic cell removal, and abnormal activation of the type 1 interferon pathway, all of which contribute to the breakdown of immune tolerance and tissue damage.2 Among the key pathological processes in SLE, vasculitis, defined as inflammatory infiltration and necrosis of blood vessel walls, is particularly significant.3,4 This includes both systemic and cutaneous vasculitis (CV), with the latter referring specifically to inflammation involving vasculature of the skin.3 Not only does it serve as a hallmark of disease progression, but it is also one of the leading causes of mortality in individuals with established SLE.3,4 The clinical presentation varies according to the vessel size and location, ranging from mild to severe complications.3 In general, skin lesions and rashes are among the most frequent dermatologic symptoms observed in patients with SLE, stemming from inflammation affecting small and medium‑sized blood vessels in the skin. CV often serves as a warning sign of potentially more severe and life‑threatening inflammatory processes occurring in vital organs of the body.5
Given its clinical significance, understanding the role of vasculitis in SLE is crucial for optimal clinical management. This vascular inflammatory process is highlighted in disease activity metrics, such as the SLE Disease Activity Inde × 2000 (SLEDAI‑2K), underscoring its importance in monitoring and managing SLE.4 Proper identification and management of vasculitis are crucial, as this condition often requires more aggressive treatment strategies to mitigate potentially fatal outcomes.6 The prevalence of SLE‑associated vasculitis has been reported to range from 11% to 36%, with small vessel vasculitis of the skin being the most commonly observed type, accounting for 82%–89% of cases.7 Ramos‑Casals et al8 identified CV in 68 of 670 patients with SLE, corresponding to a prevalence of 10.1%. However, these prevalence rates may be overestimated, and confirmation via skin biopsy could improve diagnostic precision and reduce overestimation.7 CV, often linked to systemic vasculitis or underlying connective tissue diseases such as SLE, requires prompt recognition and systematic evaluation to guide effective management and improve outcomes.9 Interestingly, in pediatric SLE patients, CV is notably prevalent, affecting 36% of cases, and is associated with unique features, such as male predominance, younger age at diagnosis, family history of SLE, conjunctivitis, low C3 levels, and presence of cytoplasmic antineutrophil cytoplasmic antibodies.10
In recent years, increasing attention has been paid to the pathophysiology of vasculitis in the course of SLE. For instance, Wu et al11 observed elevated serum levels of growth arrest–specific protein 6 in SLE patients with CV, highlighting the role of this protein in inflammatory processes and its potential as a biomarker of disease activity. Next, endothelial cell activation, reflected by increased plasma levels of tissue plasminogen activator antigen, von Willebrand factor antigen, and tissue factor, was shown to play a central role in the pathophysiology of CV in SLE, contributing to a shift from an anticoagulant to a prothrombotic state, and driving local fibrin deposition and inflammatory vascular damage.12 Furthermore, based on a study by D’Cruz et al,13 a high prevalence of antiendothelial cell antibodies in SLE patients with urticarial vasculitis highlights the potential role of these antibodies in endothelial damage and the pathogenesis of CV in this context. According to a study by Sahin et al,14 serum pentraxin‑3 levels are elevated in childhood‑onset SLE, and may serve as a significant mediator of active CV, correlating with disease activity, particularly in patients with mucocutaneous manifestations and Raynaud phenomenon.
Importantly, the diverse skin manifestations of lupus not only aid in making a diagnosis (through characteristic clinicopathological correlations) but also provide crucial prognostic and therapeutic guidance, highlighting the need for tailored treatment strategies based on specific lesion types and associated systemic risks.15 In fact, accurate diagnosis of cutaneous lupus erythematosus requires careful analysis of clinical, histological, serological, and immunofluorescence findings, particularly when dealing with lesser‑known variants and newly described features.16 Episodes of vasculitis in SLE frequently coincide with disease flares and are often accompanied by constitutional symptoms, such as fever, fatigue, and weight loss.3 Additionally, patients with vasculitis may exhibit higher rates of livedo reticularis, anemia, elevated erythrocyte sedimentation rate, and anti–Sjögren syndrome (SS) type B autoantibody positivity.3 Furthermore, overlapping conditions linked to SLE, such as SS and cryoglobulinemia, can also manifest as CV, complicating the classification and diagnostic framework of SLE‑associated vasculitis.7 Patients with CV may also report purpura, petechiae, or ulcers, particularly on the lower extremities, which consitute important diagnostic clues.7
To our knowledge, no studies to date have examined CV in a large cohort of SLE patients from Central or Eastern Europe, including Poland. Given the potential regional variations in clinical presentation and health care practices, such data may provide valuable insights into disease heterogeneity. To address this gap, we analyzed the clinical characteristics, laboratory features, and treatment outcomes of CV in a large Polish cohort of patients with SLE.
Patients and methods
Study population
We conducted a retrospective review of medical records of patients diagnosed with and treated for SLE at the University Hospital in Kraków, Poland, between 2012 and 2022. All included patients met the 2019 classification criteria for SLE defined by the European League Against Rheumatism (EULAR) and the American College of Rheumatology (ACR).17
Detailed information on the methods and key outcomes, including definitions related to SLE and comorbidities, is available in our previous publication.18 Briefly, we gathered extensive data on patient demographics (sex, age, family history of autoimmune diseases), clinical presentations, and laboratory findings. These encompassed details regarding the timing of SLE symptom onset and diagnosis, duration of the disease, associated comorbidities, miscarriages in women, treatment regimens, as well as the causes of and age at death, when relevant. The clinical manifestations evaluated included skin lesions, joint involvement, serositis, hematologic parameters, organ‑specific issues (related to the kidney, liver, nervous system, and respiratory tract), Raynaud phenomenon, and lymphadenopathy.
Next, we classified the patients into 2 distinct cohorts: those diagnosed with CV and those without such manifestations (non‑CV). The diagnosis of CV was confirmed through clinical assessment and / or histologic examination7,19; the latter was performed in 4.7% of the CV cases (3 out of 64 CV patients). The lesions manifested as punctate spots, palpable purpura, and tender erythematous plaques or macules, with or without necrosis.19 The clinical diagnosis of CV was confirmed by at least 2 physicians representing the following medical specialties: internal medicine, dermatology, clinical immunology, or rheumatology; all of them had formal training and expertise in diagnosing CV in connective tissue diseases.
Ethics
The study received approval from the Bioethics Committee of the Jagiellonian University Medical College (118.6120.21.2023). All research activities complied with the ethical standards set forth in the Declaration of Helsinki.
Laboratory analysis
We used standard laboratory methods to assess complete blood counts, lipid profiles, creatinine level, and glomerular filtration rate calculated via the Modification of Diet in Renal Disease formula. Urine analyses included 24‑hour protein excretion and sediment evaluation. Antinuclear antibody (ANA) testing was performed with indirect immunofluorescence on HEp‑2 cells, followed by an enzyme‑linked immunosorbent assay or line‑blot for specific antibodies. Anti–double‑stranded DNA (anti‑dsDNA) levels were measured using Crithidia luciliae, and complement levels were assessed via nephelometry (reference range [RR] <1:10). Hypercoagulability tests covered lupus anticoagulant (LA) and anticardiolipin (aCL)/anti–β2 glycoprotein I (aβ2GPI) antibodies (immunoglobulin [Ig] classes M and G). The RRs for aCL antibodies were 0–15 GPL for the IgG class and 0‑12 MPL for the IgM class. For aβ2GPI antibodies, the RRs were 0–20 SGU for IgG and 0–20 SMU for IgM.
Statistical analysis
Data were analyzed using TIBCO Statistica software, version 13.3 (StatSoft Inc., Tulsa, Oklahoma, United States). Categorical variables were reported as counts and percentages, with comparisons performed using the χ2 test or Fisher exact test. Continuous variables, which were found to have a non‑normal distribution (based on the Shapiro–Wilk test), were described as medians with interquartile ranges (IQRs) and compared using the Mann–Whitney test. For binary variables, odds ratios (ORs) with 95% CI were calculated. In all tests, a P value below 0.05 was assumed as significant.
Results
Demographic features
The CV and non‑CV SLE patients were similar in terms of sex distribution and age at the time of the study and at SLE onset, with a marked predominance of women in both groups. However, the CV patients had longer disease duration than the non‑CV individuals (median [IQR], 18 [12–24] vs 12 [6–12] years; P = 0.002). Regarding smoking status, the patients included in the CV group had a higher OR of representing active smokers, as compared with the non‑CV group (OR, 2.48; 95% CI, 1.30–4.73). Detailed demographic characteristics are presented in Table 1. Over the 14‑year follow‑up, 50 patients died, with no significant differences between the analyzed subgroups with respect to the mortality rate or cause of death (Supplementary material, Table S1).
Characteristics | SLE patients with CV (n = 64) | SLE patients without CV (n = 957) | P value |
Data are presented as number (percentage) or median (interquartile range).
P values <0.05 were considered significant.
Abbreviations: CV, cutaneous vasculitis; SLE, systemic lupus erythematosus | |||
Women | 56 (87.5) | 852 (89) | 0.71 |
Current age, y | 49 (40–61) | 49 (38–61) | 0.62 |
Age at SLE onset, y | 31 (22–44) | 35 (25–48) | 0.12 |
SLE duration, y | 18 (12–24) | 12 (6–20) | 0.002 |
Deaths | 6 (10.53) | 44 (5.03) | 0.14 |
Smokers | 23 (57.5) | 17 (42.5) | 0.005 |
Clinical manifestations
The prevalence of systemic symptoms across the SLE study groups is summarized in Table 2. In the CV group, the most prevalent clinical manifestations included mucocutaneous symptoms (100%), joint issues (96.88%), and constitutional symptoms (87.5%). The patients with CV often presented with multiple clinical manifestations. Specifically, they exhibited a higher incidence of constitutional symptoms (OR, 2.19; 95% CI, 1.03–4.66), including fever (OR, 1.95; 95% CI, 1.17–3.27), myalgia (OR, 2.22; 95% CI, 1.33–3.72), weight loss (OR, 1.78; 95% CI, 1.02–3.1), and lymphadenopathy (OR, 1.79; 95% CI, 1.01–3.17). Moreover, the CV group presented higher prevalence of other symptoms, including urticaria (OR, 3.28; 95% CI, 1.7–6.32), oral / nasal ulcers (OR, 2.18; 95% CI, 1.22–3.91), arthralgia (OR, 4.67; 95% CI, 1.13–19.33), and central nervous system (CNS) involvement (OR, 2.62; 95% CI, 1.27–5.4). Interestingly, there were no differences in the prevalence of kidney involvement, serositis, Raynaud phenomenon, and lupoid hepatitis. Furthermore, the number of patients with lupus nephritis exacerbation did not differ between the CV and non‑CV SLE groups (Supplementary material, Table S2).
Clinical manifestations | SLE patients with CV (n = 64) | SLE patients without CV (n = 957) | P value | ||
Data are presented as number (percentage).
P values <0.05 were considered significant.
a Leukocyte count <4000 cells/µl or diagnosis based on medical history
b Lymphocyte count <1500 cells/µl or diagnosis based on medical history
c Hemoglobin level ≤12 g/dl in women, ≤13.5 g/dl in men, or diagnosis based on medical history
d Anemia with positive direct Coombs test, anemia with decreased haptoglobin level, or diagnosis based on medical history
e Thrombocyte count <100 000 cells/µl or diagnosis based on medical history
Abbreviations: see Table 1 | |||||
Constitutional symptoms | Any type | 56 (87.5) | 729 (76.2) | 0.04 | |
Fever | 36 (57.1) | 380 (40.6) | 0.001 | ||
Fatigue / weakness | 47 (74.6) | 582 (62.1) | 0.047 | ||
Myalgia | 35 (55.6) | 337 (36) | 0.002 | ||
Weight loss | 20 (31.8) | 194 (20.7) | 0.04 | ||
Lymphadenopathy | 18 (28.1) | 168 (17.9) | 0.04 | ||
Mucocutaneus manifestations | Any type | 64 (100) | 756 (79) | <0.001 | |
Lupus malar rash | 26 (40.6) | 417 (43.6) | 0.64 | ||
Discoid rash | 6 (9.4) | 77 (8.1) | 0.71 | ||
Urticaria | 13 (20.3) | 69 (7.2) | <0.001 | ||
Alopecia | 22 (34.4) | 236 (24.7) | 0.08 | ||
Oral and / or nasal ulcers | 17 (26.6) | 136 (14.2) | 0.007 | ||
Photosensitivity | 23 (35.9) | 320 (33.5) | 0.69 | ||
Joint manifestations | Any type | 62 (96.9) | 835 (87.3) | 0.02 | |
Arthritis | 45 (71.4) | 558 (55.8) | 0.048 | ||
Arthralgia | 62 (96.9) | 830 (86.9) | 0.02 | ||
Serositis | Any type | 15 (23.4) | 198 (20.7) | 0.3 | |
Pleural effusion | 10 (15.6) | 159 (16.7) | 0.83 | ||
Pericardial effusion | 10 (15.6) | 130 (13.8) | 0.68 | ||
Pericarditis | 2 (3.1) | 38 (4) | >0.99 | ||
Hematological manifestations | Any type | 60 (93.8) | 855 (89.3) | 0.39 | |
Leucopeniaa | 43 (67.2) | 559 (59.9) | 0.25 | ||
Lymphopeniab | 34 (53.1) | 376 (39.3) | 0.04 | ||
Anemiac | 47 (73.4) | 653 (69.7) | 0.53 | ||
Hemolytic anemiad | 4 (14.3) | 84 (19.4) | 0.63 | ||
Thrombocytopeniae | 23 (35.9) | 280 (30) | 0.32 | ||
Kidney involvement | Any type | 28 (43.8) | 353 (36.9) | 0.27 | |
24‑h urinary protein excretion >0.5 g/day | 20 (33.9) | 299 (32.1) | 0.78 | ||
24‑h urinary protein excretion >3.5 g/day | 10 (17.2) | 153 (17.3) | >0.99 | ||
Urinary casts | 8 (14.3) | 133 (15.7) | 0.77 | ||
Erythrocyturia | 21 (35.6) | 228 (25.6) | 0.09 | ||
Leucocyturia | 20 (32.8) | 265 (29.3) | 0.56 | ||
Neurological signs | Any type | 14 (21.9) | 95 (9.9) | 0.003 | |
Central nervous system involvement | 10 (15.6) | 63 (6.6) | 0.007 | ||
Peripheral nervous system involvement | 5 (7.8) | 45 (4.7) | 0.27 | ||
Raynaud phenomenon | 22 (34.4) | 224 (23.4) | 0.048 | ||
Lung involvement | Any type | 10 (15.6) | 73 (7.6) | 0.02 | |
Interstitial lung disease | 5 (7.8) | 47 (4.9) | 0.31 | ||
Diffuse alveolar hemorrhage | 2 (3.1) | 9 (0.9) | 0.15 | ||
Pulmonary hypertension | 4 (6.3) | 28 (3) | 0.14 | ||
Lupoid hepatitis | 3 (4.7) | 50 (5.2) | >0.99 | ||
Comorbidities
The prevalence of comorbidities was similar between the analyzed groups (Table 3), except for a higher rate of heart failure (HF; OR, 3.55; 95% CI, 1.65–7.67) among the CV patients. Consistent with the lack of differences in the prevalence of hypercholesterolemia or myocardial infarction, the CV and non‑CV groups also did not differ in the frequency of statin treatment (Table 4). Furthermore, there were no significant differences with respect to the occurrence of arterial thromboembolic events or venous thromboembolism (VTE).
Comorbidity | SLE patients with CV (n = 64) | SLE patients without CV (n = 957) | P value |
Data are presented as number (percentage).
P values <0.05 were considered significant.
Abbreviations: see Table 1 | |||
Hypothyroidism | 13 (20.3) | 234 (24.5) | 0.45 |
Hyperthyroidism | 3 (4.7) | 51 (5.3) | >0.99 |
Hypertension | 35 (54.7) | 503 (52.6) | 0.75 |
Diabetes mellitus | 7 (10.9) | 97 (10.1) | 0.84 |
Heart failure | 9 (14.1) | 42 (4.4) | <0.001 |
Hypercholesterolemia | 32 (50) | 450 (47.1) | 0.66 |
Atrial fibrillation | 2 (3.1) | 36 (3.8) | >0.99 |
Peripheral artery disease | 4 (6.3) | 52 (5.4) | 0.77 |
End‑stage kidney disease | 1 (1.6) | 26 (2.7) | >0.99 |
Kidney transplant | 0 | 9 (0.9) | >0.99 |
Malignant tumor | 10 (15.6) | 98 (10.3) | 0.18 |
Arterial thromboembolic episode | 9 (14.1) | 95 (9.9) | 0.29 |
Stroke | 6 (9.4) | 74 (7.7) | 0.64 |
Transient ischemic attack | 3 (4.7) | 13 (1.4) | 0.07 |
Myocardial infarction | 5 (7.8) | 52 (5.4) | 0.42 |
Other arterial thromboembolic episodes | 3 (4.7) | 19 (2) | 0.16 |
Venous thromboembolism | 14 (21.9) | 169 (17.7) | 0.39 |
Deep vein thrombosis | 12 (18.8) | 141 (14.8) | 0.39 |
Pulmonary embolism | 4 (6.3) | 39 (4.1) | 0.34 |
Deep vein thrombosis and pulmonary embolism | 2 (3.1) | 21 (2.2) | 0.65 |
Other venous thomboembolic episodes | 1 (1.6) | 18 (1.9) | >0.99 |
Miscarriage | 8 (14.5) | 87 (10.2) | 0.3 |
Treatment | SLE patients with CV (n = 64) | SLE patients without CV (n = 957) | P value |
Data are presented as number (percentage).
P values <0.05 were considered significant.
Abbreviations: iv, intravenous; others, see Table 1 | |||
Glucocorticoids (oral / iv) | 63 (98.4) | 899 (94.2) | 0.25 |
Chloroquine or hydroxychloroquine | 53 (82.8) | 721 (75.7) | 0.19 |
Azathioprine | 33 (51.6) | 355 (37.5) | 0.02 |
Cyclosporin | 6 (9.4) | 74 (7.8) | 0.66 |
Methotrexate | 18 (28.13) | 180 (18.81) | 0.11 |
Belimumab | 6 (9.4) | 35 (3.7) | 0.03 |
Mycophenolate mofetil | 17 (26.6) | 285 (30.2) | 0.54 |
Cyclophosphamide | 26 (40.6) | 260 (27.5) | 0.02 |
Rituximab | 1 (1.6) | 29 (3.1) | >0.99 |
Immunoglobulins | 2 (3.1) | 26 (2.8) | 0.7 |
Plasmapheresis | 1 (1.6) | 30 (3.2) | 0.72 |
Anifrolumab | 0 | 10 (1.1) | >0.99 |
Statins | 32 (50) | 499 (52.1) | 0.74 |
Anticoagulants | 17 (26.6) | 210 (21.9) | 0.39 |
Autoantibody profiles
In the SLE patients with CV symptoms, anti‑SS type A (anti‑SSA; OR, 2.07; 95% CI, 1.13–3.76) and antiribonucleoprotein (anti‑RNP) antibodies (OR, 2.11; 95% CI, 1.21–3.68) were more prevalent than in the individuals without skin vasculitis. Interestingly, rheumatoid factor presence was observed more often in the CV group (OR, 2.39; 95% CI, 1.21–4.72). No significant differences between the analyzed groups were observed with respect to other ANAs, antiphospholipid antibodies (APLAs), or the presence of antiphospholipid syndrome (APS), as assessed based on the 2023 ACR/EULAR classification criteria.20 Additionally, there were no intergroup differences with respect to the frequency of anticoagulant treatment (Table 4). Detailed information on the antibody profile is provided in Table 5.
Parameter | SLE patients with CV (n = 64) | SLE patients without CV (n = 957) | P value |
Data are presented as number (percentage).
P values <0.05 were considered significant.
a Diagnosed according to the 2023 ACR/EULAR classification criteria
Abbreviations: dsDNA, double‑stranded DNA; Ig, immunoglobulin; RNP, ribonucleoprotein; SSA, Sjögren syndrome type A; SSB, Sjögren syndrome type B; others, see Table 1 | |||
Anti‑SSA antibodies | 45 (75) | 526 (59.2) | 0.02 |
Anti‑SSB antibodies | 23 (38.3) | 263 (29.6) | 0.15 |
Antihistone antibodies | 15 (25) | 236 (26.6) | 0.79 |
Antinucleosome antibodies | 16 (26.7) | 295 (33.2) | 0.3 |
Anti‑Smith antibodies | 8 (13.3) | 106 (12) | 0.76 |
Anti‑RNP antibodies | 21 (35) | 180 (20.3) | 0.007 |
Anti‑dsDNA antibodies | 16 (26.7) | 334 (37.8) | 0.08 |
Lupus anticoagulant | 12 (23.5) | 193 (29.7) | 0.35 |
Anticardiolipin IgG and / or IgM | 31 (53.4) | 410 (56.8) | 0.62 |
Anti-β2 glycoprotein I IgG and / or IgM | 11 (24.4) | 156 (26.9) | 0.73 |
Antiphospholipid syndromea | 11 (23.4) | 169 (26.2) | 0.67 |
Immunosuppressive treatment
Next, we analyzed various approaches to immunosuppressive treatment (Table 4). Glucocorticosteroids were the most frequently administered drugs among both the CV and non‑CV SLE patients (98.44% and 94.23%, respectively; P = 0.25). The individuals with CV, as compared with the non‑CV ones, were more often prescribed an aggressive immunosuppressive regimen involving azathioprine (OR, 1.78; 95% CI, 1.07–2.95), belimumab (OR, 2.68; 95% CI, 1.09–6.64), and cyclophosphamide (OR, 1.8; 95% CI, 1.07–3.03). There were no significant differences between the groups in the usage of other immunosuppressants (Table 4).
Discussion
This study presented clinical and laboratory characteristics of a large cohort of SLE patients treated at a single center, with a focus on individuals with CV. Our findings show that the presence of skin vasculitis impacts the clinical course of the disease. The variations in the occurrence of CV in SLE not only underscore the heterogeneous nature of the disease but also highlight critical pathways that may inform more personalized and targeted clinical interventions, which are highly recommended nowadays.22 To our best knowledge, this study describes one of the largest Polish cohorts of SLE patients in the literature, comparing various aspects of the disease based on the presence of skin vasculitis.
CV changes are not prevalent in SLE.21 In our study, only 6.27% of the patients presented with such manifestations. Our findings are in line with those reported in a multicenter cohort analyzed by Breillat et al.7 The authors concluded that CV in SLE was relatively rare, with a significant proportion of patients exhibiting no signs of active SLE at the time of diagnosis, emphasizing the need to rule out other potential causes of vasculitis. Importantly, Breillat et al7 investigated vasculitis in 39 SLE patients, with diagnoses confirmed by histologic examination in all of them—an aspect that significantly strengthens the validity of their findings in contrast to our study. However, in a study by Kallas et al,19 CV was found to be relatively frequent in SLE patients, with almost one‑fifth (17.4%) of the analyzed cohort presenting with skin vasculitis. These discrepancies might be related to different definitions of CV in SLE, and the findings should be interpreted with caution.
Our demographic analysis showed that the CV and non‑CV groups were comparable in terms of sex distribution and age at the time of the study. However, the patients with CV were younger at the onset of SLE, which aligns with previous reports suggesting that early‑onset lupus may be associated with more severe disease manifestations and complications.23,24 The mortality rate was similar in both groups. It has been shown that skin manifestation in SLE may differ according to the age at the onset of the disease. Based on a study by Chottawornsak et al,25 in juvenile‑onset SLE, acute cutaneous lupus erythematosus and nonscarring alopecia were associated with an increased risk of arthralgia, whereas in adult‑onset SLE, CV correlated with myositis, highlighting the distinct systemic involvements based on the age of onset.
CV in SLE is a significant manifestation, increasing the risk of complications and mortality.26 The pathogenesis involves immune complex deposition in blood vessel walls, triggering inflammation and complement activation, leading to vascular damage.26 Additionally, the presence of APLAs in SLE elevates the risk of thrombosis, worsening vascular complications and organ damage, especially involving the kidneys and cardiovascular system.26 Chronic vascular inflammation might cause irreversible organ damage and increase susceptibility to infections due to immunosuppressive treatments used in managing SLE.27 Our results emphasize that the primary cause of death in the CV group was linked to SLE exacerbation. Therefore, alongside CV, other vasculitic changes, such as those occurring in the CNS, should be considered as potential risk factors for death.
The clinical manifestations of SLE were markedly more severe in the CV group, with higher incidence of constitutional symptoms, mucocutaneous manifestations, and joint issues. Specifically, constitutional symptoms, such as fever, myalgia, weight loss, and lymphadenopathy, were more common in the CV group, as were mucocutaneous manifestations, including urticaria and oral / nasal ulcers. Interestingly, clinical presentations such as myositis and hematological manifestations have been suggested as predictors of CV development.19 In our analysis, no differences were found regarding the prevalence of kidney involvement. Thus, our results are in line with those reported by Shinjo et al,28 who showed that among the SLE patients with CV, the frequency of renal and CNS involvement was similar to that observed in the non‑CV individuals. In contrast, Kallas et al19 highlighted that juvenile SLE patients presenting with CV alone may exhibit more granular casts in the urine sediment. Similarly, adult patients with CV may be more likely to develop renal symptoms in the course of SLE.19 However, in general, increased frequency of kidney involvement, including urinary casts, erythrocyturia, and leucocyturia, is a critical complication in SLE, and translates to worse long‑term prognosis.29 Interestingly, we found no significant differences between the CV and non‑CV groups in terms of lung involvement, serositis, or lupoid hepatitis. Hence, reports of gastrointestinal and pulmonary vasculitis in patients with SLE are uncommon in the literature.22
Of note, the CV group showed a higher prevalence of HF, a serious comorbidity that could contribute to their mortality rate. It seems that monitoring cardiovascular health in patients with SLE and CV is crucial, as HF may represent a significant complication that warrants early intervention and more aggressive management.30 Furthermore, a recent study by Saleh et al31 pointed out the importance of monitoring systemic features, such as Raynaud phenomenon and cardiovascular involvement (both of which are predictive of CV) to facilitate timely intervention. In our study, Raynaud phenomenon was relatively frequent and occurred at a similar rate in both CV and non‑CV groups. Skin microvascular dysfunction in SLE patients, even without evident cardiovascular disease, may indicate early vascular damage, warranting further investigation.32 On the other hand, no significant differences between the CV and non‑CV patients were observed with respect to the frequency of arterial thromboembolic events or VTE occurrence, which might reflect similar thromboembolic risk profiles in both groups. Indeed, CV in SLE is important; as recently indicated by Saleh et al,31 patients with CV are more likely to develop at least 1 item of organ damage.
In our study, the prevalence of APS diagnosis was similar in the CV and non‑CV groups. A lack of differences in anticoagulant treatment further supports these findings. Classic APLAs (LA, aCL, and aβ2GPI of the IgG or IgM isotypes) are not always detectable in patients with clinical manifestations of APS.33,34 In such cases, particularly when unexplained thromboembolic events or obstetric complications suggestive of APS are present, the possibility of noncriteria antibody presence should be considered.33 These include IgA isotypes of aCL and aβ2GPI, antiphosphatidylserine‑prothrombin antibodies, and antibodies against domain I of β2GPI.33 Testing for these noncriteria APLAs may be especially valuable in the cases of suspected seronegative APS, or when standard laboratory criteria yield inconclusive results despite clear clinical symptoms.33
Early recognition of CV is essential, as it can serve as a key indicator of systemic involvement, particularly in autoimmune diseases such as SLE, and prompt appropriate diagnostic workup and treatment.35 Indeed, presence of CV in SLE is associated with an increased risk of organ damage.19 Importantly, in a study by Lun et al,36 vasculitis was identified as a significant risk factor for the development of pulmonary arterial hypertension in patients with SLE, with a higher risk observed in those with combined vasculitis (OR, 1.50; 95% CI, 1.08–2.07; P <0.05).
SLE is a complex immune‑mediated disease associated with production of a wide variety of autoantibodies. In our study, the antibody profile of SLE patients with CV showed distinct differences in comparison with that of the non‑CV individuals, with anti‑SSA and anti‑RNP antibodies being more common in the CV group. These findings are consistent with previous studies reporting a strong association between certain autoantibodies, such as anti‑SSA and anti‑RNP, and more severe forms of SLE. While we observed no significant differences in other autoimmune markers, such as anti‑dsDNA or APLAs, the higher prevalence of anti‑SSA and anti‑RNP antibodies in the CV group could be indicative of a specific immunological subtype of SLE that is more prone to developing CV. Saleh et al31 observed a strong association of CV with anti‑Smith antibodies, anti‑dsDNA, and hypocomplementemia, which might highlight the role of autoantibodies and complement activation in its pathogenesis, reinforcing the need for serological monitoring in high‑risk SLE patients. Moreover, anti‑Ro antibody might serve as an independent serological marker for the development of CV in patients with SLE.37 Recent research has focused on finding new antibodies that are potentially related to SLE manifestations and might explain its complex pathogenesis. For example, the findings from a study by Shi et al38 underscore the importance of exploring nonroutinely measured autoantibodies, such as antiacidic ribosomal protein P0 and anti–galectin 3, which may play a key role in skin lesion development. These antibodies were found to correlate with lupus‑specific CV and other clinical manifestations, such as leukopenia and C3 deficiency, suggesting that they could serve as potential biomarkers of disease activity and provide new insights into the immune mechanisms underlying SLE‑related organ damage. In a study by Shinjo et al,28 patients with SLE and CV were characterized by a higher prevalence of Raynaud phenomenon and anti–ribosomal P antibodies.
It has been postulated that CV might be related to disease activity in SLE.19 We did not collect data on disease activity (such as the SLEDAI score or complement levels) in our cohort. Nevertheless, Gamal et al39 showed that higher SLEDAI scores at the disease onset were linked to the development of vasculitis, suggesting that high disease activity may drive vascular complications. In a study by Gheita et al,26 hypocomplementemia, specifically low C3 and C4 levels, was found to be closely associated with CV in SLE, highlighting the role of complement activation in its pathogenesis. Additionally, the authors observed significant links between CV and lupus nephritis, cardiovascular manifestations, and SS, suggesting that CV may indicate more severe systemic involvement in SLE.
Cutaneous manifestations in general, such as subacute cutaneous lupus erythematosus and malar rash, are not only common in SLE patients but also closely correlate with higher disease activity and systemic involvement.40 Clinicians should be particularly vigilant when observing these lesions, as they may signal more severe disease progression and an increased risk of complications, such as vasculitis, nephritis, and skin infections, warranting more frequent monitoring and aggressive management. Importantly, in a study by Olbrich et al,41 cutaneous lupus erythematosus, particularly its chronic discoid subtype, was found to be associated with an increased risk of various cardiac and vascular diseases, including thromboembolic events, such as pulmonary embolism, cerebral infarction, and acute myocardial infarction. Heil et al42 reported that lupus erythematosus skin disease was found to be common among SLE patients, and was associated with photosensitivity and presence of specific autoantibodies, but it did not significantly impact the severity of SLE sequelae, disease flares, or overall survival.
Regarding treatment, SLE patients with CV were more frequently prescribed aggressive immunosuppressive therapies, including azathioprine, cyclophosphamide, and belimumab. This reflects the more severe nature of their disease, as these treatments are typically used for patients with active or refractory disease. The fact that the CV group received a more intensive immunosuppressive regimen emphasizes the need for tailored treatment strategies for patients with this complication. However, it is also worth noting that both groups were similarly often treated with glucocorticoids, which remain a cornerstone of SLE therapy. Overall, our study highlights the more severe clinical course and worse prognosis of SLE patients with CV. These patients exhibit more frequent and severe systemic manifestations, higher comorbidity rates, and distinct antibody profiles, as compared with those without CV. Timely and targeted treatment of vascular changes in SLE is crucial, with antimalarials being the first‑line option for CV. More severe cases with visceral organ involvement may require aggressive immunosuppressive therapy, including corticosteroids, cyclophosphamide, and emerging biologics, such as like rituximab and belimumab for refractory cases.3 In fact, the marked decline in CV incidence over the 20‑year follow‑up, as reported by Saleh et al,31 suggests improvements in SLE management, particularly through the widespread use of antimalarials and immunosuppressants.
Limitations
Some limitations of our study need to be acknowledged. Firstly, its retrospective design may have led to inherent biases in both data collection and patient selection. Next, only a few patients had CV confirmed through histologic examination, which increases the risk of misdiagnosing skin changes. However, clinical diagnosis of CV was confirmed by at least 2 doctors trained in diagnosing CV in connective tissue diseases. Furthermore, we did not collect data on body mass index. Also, the single‑center design of the study may restrict the applicability of the findings to broader populations. Another limitation is the absence of patient‑reported outcomes, such as quality‑of‑life questionnaires, which could enable a more comprehensive evaluation of patient well‑being, including the effects of the disease and treatment approach. Some of the observed associations might be coincidental rather than indicative of causation. Furthermore, due to the retrospective nature of the study, the use of disease activity indices, such as the British Isles Lupus Assessment Group Index) or SLEDAI, along with the assessment of C3 and C4 levels, was not feasible. This limitation stemmed from the absence of specific data required for score calculation at potential follow‑up time points and the variability in follow‑up duration. Unfortunately, skin lesion photographs were not available due to the retrospective nature of the study.
Conclusions
SLE patients with CV are at an increased risk of organ damage (especially the cardiovascular system), emphasizing the importance of early detection and aggressive management to mitigate long‑term complications. Importantly, when CV is observed in SLE patients, it is crucial to look for other types of organ / system‑related vasculitis, especially those affecting life‑important organs, such as the heart and CNS. Presence of CV is associated with more aggressive disease progression, and a greater need for intensive immunosuppressive therapies. Thus, early recognition and targeted management of CV are essential for improving long‑term patient outcomes.
- Hoi A, Igel T, Mok CC, Arnaud L. Systemic lupus erythematosus. Lancet. 2024; 403: 2326‑2338. | Crossref
- Tsokos GC, Lo MS, Reis PC, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016; 12: 716‑730. | Crossref
- Barile‑Fabris L, Hernández‑Cabrera MF, Barragan‑Garfias JA. Vasculitis in systemic lupus erythematosus. Curr Rheumatol Rep. 2014; 16: 440. | Crossref
- Gladman DD, Ibañez D, Urowitz MB. Systemic lupus erythematosus disease activity inde × 2000. J Rheumatol. 2002; 29: 288‑291. | Crossref
- Pullen RL. Managing cutaneous vasculitis in a patient with lupus erythematosus. Dermatol Nurs. 2007; 19: 21‑26. | Crossref
SUPPLEMENTARY MATERIAL
ARTICLE INFORMATION