Genetic architecture of dilated cardiomyopathy in Poland: variant distribution, clinical characteristics, and prognosis
Key words: dilated cardiomyopathy, genetics, next-generation sequencing, prognosis

 CC BY 4.0
CC BY 4.0
Genetic architecture of dilated cardiomyopathy in Poland: variant distribution, clinical characteristics, and prognosis
Introduction: Genetic variants are the leading cause of dilated cardiomyopathy (DCM). Data on genetic testing in DCM from Central European populations are scarce.
Objectives: We sought to determine the genetic architecture of DCM in Poland and to assess its influence on patient clinical characteristics and prognosis.
Patients and methods: The study included 280 DCM patients (mean [SD] age, 39 [13] y; 68% men) who underwent next‑generation sequencing or in whom disease‑causing variants were identified using single‑gene testing.
Results: DCM‑related variants were identified in 46% of the patients. Variants in titin (TTN) and lamin A/C (LMNA) genes were the most frequent (18% and 8% of the study cohort, respectively). Other genes with variant frequency equal to or above 2% included desmoplakin (DSP), myosin heavy chain 7 (MYH7), sodium voltage‑gated channel α subunit 5 (SCN5A), filamin C (FLNC), BCL2‑associated athanogene 3 (BAG3), and dystrophin (DMD). Positive family history, atrioventricular block, or atrial arrhythmias were found more often in the causative variant carriers, whereas the frequency of left bundle branch block or hypertension was lower. Severe DCM and a composite end point (cardiovascular death, heart transplantation, left ventricular assist device implantation) occurred with similar frequency in gene‑negative DCM and TTN-related DCM, whereas the prognosis was worse in the remaining variant carriers. The risks of severe DCM and the composite end point were 2.4- and 3‑fold higher, respectively, for LMNA-related DCM and 2.3- and 2‑fold higher for variants in the other genes.
Conclusions: The distribution of causative genetic variants in Polish DCM patients is similar to that in Western European cohorts. The presence of the causative variants in the genes other than TTN is associated with a poorer prognosis.
What's new?
This is the first Central European study to assess the prevalence of genetic background in a large cohort of patients with dilated cardiomyopathy (DCM), taking into account pathogenic and likely pathogenic variants according to the American College of Medical Genetics and Genomics criteria in genes with at least moderate level of evidence for association with DCM. We found DCM‑related genetic variants in almost half of the patients, with titin (TTN) variants identified most frequently. We found a generally similar variant distribution as in Western Europe, however, with a higher frequency of lamin A/C (LMNA) variants and a low frequency of RNA‑binding motif protein 20 (RBM20) variants. The prognosis in the TTN variant carriers was similar to that in the gene‑negative DCM, whereas variants in other DCM‑related genes were associated with a significantly worse outcome. Genetic testing in DCM is essential to optimally assess prognosis, guide therapy, and identify relatives at risk.
Introduction
Dilated cardiomyopathy (DCM) is a disease of the cardiac muscle characterized by left ventricular (LV) dilation and decreased contractile function that cannot be explained solely by abnormal loading conditions or coronary artery disease.1,2 It can lead to heart failure (HF) and predispose to arrhythmias, and it is a significant cause of morbidity and mortality, especially in young people.1
The etiology of DCM is heterogeneous, with a significant role of genetic factors.2-4 Although genetic determinants of DCM are complex and not fully understood,5-8 in many families, DCM can be explained with a Mendelian, monogenic model. Rare, potentially disease‑causing genetic variants are found with a variable frequency estimated at 15%–40% of DCM cases.9-12
The advent of next‑generation sequencing (NGS) revolutionized genetic testing by enabling accurate and rapid DNA screening. This has facilitated obtaining genetic diagnoses in large cohorts of patients, and investigating how the genetic background influences the course of the disease. Several dozen genes, encoding proteins of various cardiomyocyte compartments, have been associated with the development of DCM. For 20 of them, the evidence for causal significance has been considered sufficient, while the role of the remaining ones in the etiopathogenesis of DCM requires further investigation.1,8 It has also been shown that the course of cardiomyopathy can vary depending on the underlying genetic background.10-14 At present, clinical guidelines recommend genetic diagnosis as part of the management of DCM patients and screening their families.1,15
The genetic background of DCM may vary across different populations.4,9 Most data on the genetic landscape of DCM come from centers in Western Europe and the United States (US), while data on Central European populations are scarce, with a single recent report on the results of genetic testing of DCM patients from southeastern Poland.16
We aimed to summarize the results of genetic testing of DCM patients performed as part of scientific projects carried out between 2012 and 2021 at the National Institute of Cardiology, Warsaw, Poland, and ascertain how they influence clinical characteristics and prognosis.
Patients and methods
Study design
We included adult unrelated patients with DCM who underwent genetic testing by NGS between 2012 and 2021. We also included patients who did not have LV dilation, provided that all other criteria for DCM were met, as well as patients with DCM in whom NGS testing was not performed due to the detection of a causative genetic variant by another method (mainly Sanger sequencing). In total, the group consisted of 280 individuals. The patients came from all over Poland, however, their geographic distribution was not even. Patients living in Mazowieckie and neighboring voivodeships predominated, while participants from distant regions were less numerous.
DNA sequencing and variant analysis
NGS was performed using different panels (details in Table 1 and Supplementary material, Tables S1 and S2). In 22 patients, pathogenic or likely pathogenic variants were detected by single‑gene testing, most often by direct Sanger sequencing of coding exons of lamin A/C (LMNA), BCL2‑associated athanogene 3 (BAG3), phospholamban (PLN), or sodium voltage‑gated channel α subunit 5 (SCN5A) including flanking intronic regions, less often by real‑time polymerase chain reaction methods or multiplex ligation‑dependent probe amplification.17-20
Gene | Protein | Compartment / function | Mode of inheritance | Association with DCM | Genetic panels | |||
ClinGen8 | 2023 ESC guidelines1 | CP35 (n = 7) | TSC (n = 83) | TSO/WES (n = 168) | ||||
Abbreviations: AD, autosomal dominant; CP35, custom panel of 35 cardiomyopathy‑related genes; DCM, dilated cardiomiopathy; ESC, European Society of Cardiology; M, moderate; S, strong; TSC, TrueSight Cardio; TSO, TrueSight One; WES, whole‑exome sequencing; XD, sex‑linked dominant | ||||||||
ACTC1 | Actin α | Motor sarcomeric | AD | M | M | + | + | + |
ACTN2 | Actinin α2 | Cytoskeleton | AD | M | M | + | + | + |
BAG3 | BCL2‑associated athanogene 3 | Co‑chaperone | AD | S | S | + | + | + |
DES | Desmin | Cytoskeleton | AD | S | S | + | + | + |
DMD | Dystrophin | Cytoskeleton | XD | – | S | – | + | + |
DSP | Desmoplakin | Desmosome | AD | S | S | – | + | + |
FLNC | Filamin C | Cytoskeleton | AD | S | S | – | – | + |
JPH2 | Junctophilin 2 | Junctional membrane | AD | M | M | – | + | + |
LMNA | Lamin A/C | Nuclear envelope | AD | S | S | + | + | + |
MYH7 | Myosin heavy chain 7 | Motor sarcomeric | AD | S | S | + | + | + |
NEXN | Nexillin F‑actin–binding protein | Cytoskeleton | AD | M | M | – | + | + |
PLN | Phospholamban | Sarcoplasmatic reticulum | AD | S | S | + | + | + |
RBM20 | RNA‑binding motif protein 20 | Nucleus / RNA binding | AD | S | S | + | + | + |
SCN5A | Sodium voltage‑gated channel α subunit 5 | Ion channel | AD | S | S | + | + | + |
TNNC1 | Troponin C1 | Motor sarcomeric | AD | S | S | + | + | + |
TNNI3 | Troponin I3 | Motor sarcomeric | AD | M | M | + | + | + |
TNNT2 | Troponin T2 | Motor sarcomeric | AD | S | S | + | + | + |
TPM1 | Tropomyosin 1 | Motor sarcomeric | AD | M | M | + | + | + |
TTN | Titin | Sarcomere | AD | S | S | + | + | + |
VCL | Vinculin | Cytoskeleton | AD | M | M | + | + | + |
The identified rare variants in cardiomyopathy‑related genes were classified according to the American College of Medical Genetics and Genomics (ACMG) criteria using the online tool GeneBe (https://genebe.net/; classification valid as of February 2025).21-23 They were considered causative if they were pathogenic (P) or likely pathogenic (LP) and were found in 1 of the 20 genes with strong or moderate evidence of causative relationship with DCM (hereinafter referred to as DCM‑related genes, listed in Table 1).1,8 Genes with limited or disputed association with DCM were not included.
All causative variants identified with NGS were confirmed with Sanger sequencing.
We excluded 3 patients with homozygous / compound heterozygous variants in the genes with disputed evidence for association with DCM whose cases had been published previously as genetic cardiomyopathy: 1 with a homozygous variant in the nebulin‑related anchoring protein gene,24 another with variants in the 1,4-α-glucan branching enzyme 1 gene,25 and the last with compound heterozygous variants in the tripeptidyl peptidase 1 gene.26
In order to compare clinical characteristics and prognosis, the patients with gene‑positive DCM were divided into 3 groups of carriers of P/LP variants in: the titin (TTN) gene (group 1), the LMNA gene (group 2), and other genes (group 3). The patients with more than 1 P/LP variant were not included in either of these groups.
Clinical assessment
Medical data of all participants were retrospectively collected, including baseline clinical information at the time of the initial visit, prior medical records, and follow‑up information. We analyzed the baseline data, comprising medical and family history, 12‑lead electrocardiography (ECG), echocardiography, comorbidities, and current treatment. All records were reviewed for the occurrence of major events, such as death, heart transplantation (HTx), implantation of a LV assist device (LVAD), implantable cardioverter‑defibrillator (ICD), cardiac resynchronization therapy (CRT), pacemaker implantation, sudden cardiac arrest (SCA), or sustained ventricular tachycardia (VT).
Two composite end points were defined: severe DCM (definition in the next section) and a composite end point of cardiovascular death, HTx, or LVAD implantation.
Definitions
The diagnosis of DCM was made when the LV end‑diastolic diameter (LVEDD) exceeded 112% of the predicted value, corrected for age and body surface area according to the Henry’s formula, and when the LV ejection fraction (LVEF) was below 50%.
DCM was considered gene‑positive if P/LP variants were identified in 1 of the DCM‑related genes (Table 1).
First‑degree AVB was defined by a PR interval exceeding 200 ms on standard 12‑lead ECG. High‑degree AVB included type II second‑degree or third‑degree AVB. Atrial arrhythmias included atrial fibrillation, flutter, and paroxysmal atrial tachycardia lasting at least 30 seconds.
HF was recognized when typical symptoms were present, and was staged using the New York Heart Association (NYHA) classification (classes 1–4).
We labelled cardiomyopathy as an acute onset disease if it was diagnosed during acute hospitalization due to symptoms of HF requiring inotropic agent infusion or due to myocarditis‑like symptoms (chest pain, troponin level elevation).
SCA was defined as occurring within 1 hour of the onset of acute symptoms and reversed with cardiopulmonary resuscitation.
We used the term “severe DCM” to mark advanced, symptomatic LV systolic dysfunction (LVEF <35%) persisting despite optimal medical treatment (and equivalent to the classic criteria for ICD/CRT implantation) or the occurrence of life‑threatening events. We confirmed it when one of the following occurred first: CRT/ICD implantation (persisting advanced LV dysfunction), CV death, HTx, LVAD implantation, SCA, or hemodynamically unstable VT (life‑threatening events).
DCM was considered familial if at least 1 first- or second‑degree relative was diagnosed with DCM. A family history of sudden cardiac death (SCD) was considered positive if at least 1 first‑degree relative had died suddenly before the age of 50 years.
Ethics
The study followed the Declaration of Helsinki, and was approved by the Local Bioethics Committee at the National Institute of Cardiology, Warsaw, Poland (IK.N.PIA.002.39.2153/25). All patients signed informed consent to participate in the study.
Statistical analysis
All results for categorical variables are presented as counts and percentages and for continuous variables as mean and SD or median and interquartile range (IQR). The χ2 independence or Fisher exact test was used to compare the categorical variables. The differences between continuous variables were tested with the t test (for 2 independent samples, normally distributed data). Significance of differences between the means in several groups was analyzed with one‑way analysis of variance with the post hoc Tukey–Kramer test for multiple comparisons.
Survival curves were constructed using the Kaplan–Meier method. Homogeneity of the curves was verified by the log‑rank test. The multiple‑comparison method for adjusting P values of the paired tests was based on the Tukey studentized range test with Kramer approximation. Hazard ratios (HRs) with 95% CIs were calculated using the Cox proportional hazards model. The proportionality of risk assumptions was checked based on weighted Schoenfeld residuals. In survival models, the variable defining the time to the first event or the censoring time was the patient’s age. In patients without an event, the follow‑up period extended to the most recent evaluation before November 30, 2024. A P value below 0.05 was considered significant.
All hypotheses were 2‑tailed with a 0.05 type I error.
All statistical analyses were performed using SAS statistical software, version 9.4 (SAS, Cary, North Carolina, United States).
Results
Molecular findings in the study cohort
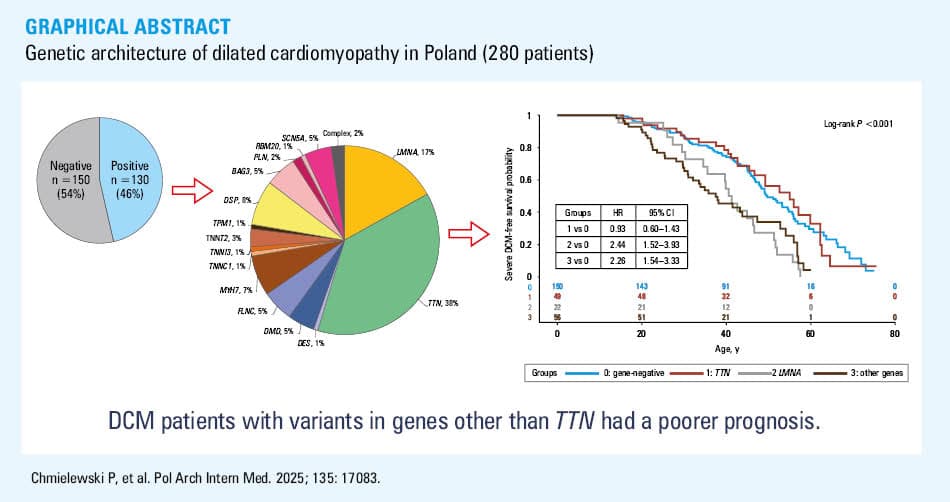
The study cohort included 280 unrelated DCM patients. NGS was performed in 258 individuals, and it was waived in 22 cases in whom a causative genetic variant was identified by another method (14 carriers of LMNA variants, 6 of BAG3 variants, 1 of PLN, and 1 of SCN5A variant). In total, 121 different P/LP variants in one of the DCM‑related genes were identified in 130 patients (46%; Figure 1), of whom 3 carried 2 variants and 127 carried a single variant (all variants are listed in Supplementary material, Tables S3 and S4). Twenty two of these variants have not been reported in the literature so far, 15 of them have not been entered into the ClinVar database. All identified variants were heterozygous except for dystrophin (DMD) hemizygous variants in men. Ten P/LP variants were found in more than 1 patient.
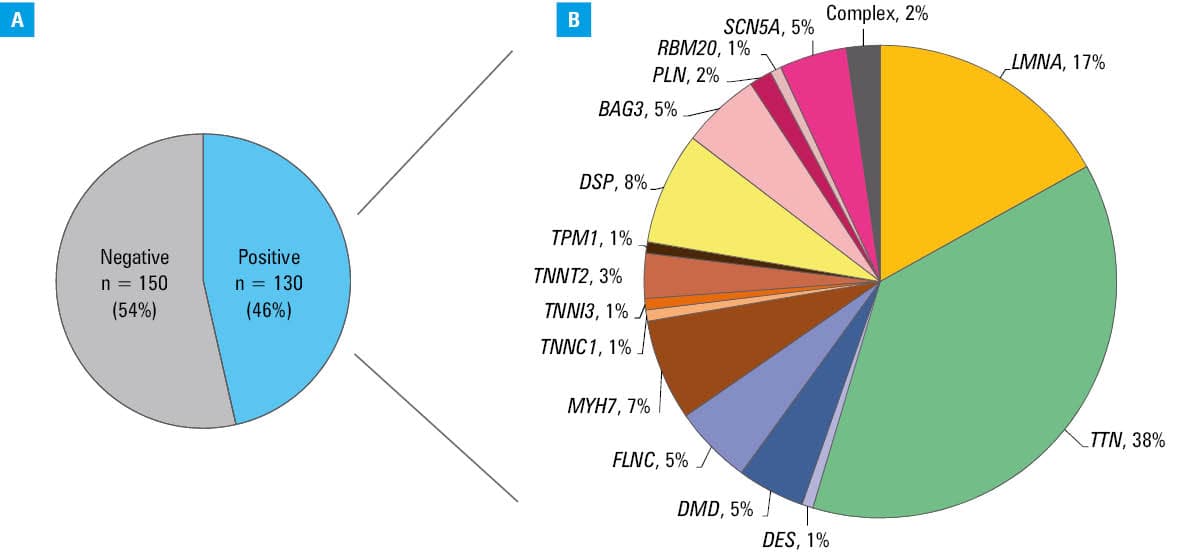
Among the patients with identified P/LP variants, as many as 39% (17% of all studied patients) had variants in the TTN gene (49 in TTN only and 2 also in another DCM‑related gene). The second most frequently affected gene was LMNA, accountable for 8% of all DCM cases and 17% of gene‑positive DCM. Other genes in which variants were identified at a frequency exceeding 2% were: desmoplakin (DSP), myosin heavy chain 7 (MYH7), SCN5A, filamin C (FLNC), BAG3, and DMD. Detailed distribution of the P/LP variants in DCM‑related genes is presented in Table 2.
Gene | N | % of all DCM patients | Gene | N | % of all DCM patients |
Abbreviations: N, number of patients with causative variants; others, see Table 1 | |||||
ACTC1 | 1 | 0.4 | PLN | 2 | 0.7 |
BAG3 | 7 | 2.5 | RBM20 | 1 | 0.4 |
DES | 1 | 0.4 | SCN5A | 8 | 2.9 |
DMD | 6 | 2.1 | TNNC1 | 1 | 0.4 |
DSP | 10 | 3.6 | TNNI3 | 1 | 0.4 |
FLNC | 7 | 2.5 | TNNT2 | 4 | 1.4 |
LMNA | 22 | 7.9 | TPM1 | 1 | 0.4 |
MYH7 | 10 | 3.6 | TTN | 51 | 18.2 |
Clinical characteristics of the study population
Clinical characteristics of the study population at baseline evaluation are listed in Table 3. The mean (SD) age of the DCM patients was 39 (13) years, 68% were men. As many as 54% of them had familial form of DCM. In 19%, the disease had an acute onset.
Variable | All (n = 280) | Group 0 (gene--negative; n = 150) | Gene‑positive (n = 130) | P value (– vs +) | Group 1 (TTN; n = 49) | Group 2 (LMNA; n = 22) | Group 3 (other genes; n = 56) | P value (0 vs 1 vs 2 vs 3) |
Data are presented as mean (SD) or number and percentage.
Abbreviations: ACEI, angiotensin‑converting enzyme inhibitor; AH, arterial hypertension; ARB, angiotensin receptor blocker; ARNI, angiotensin receptor / neprilysin inhibitor; AV block, atrioventricular block; CAD, coronary artery disease; CK, creatine kinase; CRT, cardiac resynchronization therapy; HF, heart failure; ICD, implantable cardioverter defibrillator; LBBB, left bundle branch block; LVEDD, left ventricular end‑diastolic dimension; LVEF, left ventricular ejection fraction; MRA, mineralocorticoid receptor antagonist; NYHA class, New York Heart Association functional class; PM, pacemaker; ppx, prophylaxis; SCA, sudden cardiac arrest; SCD, sudden cardiac death; others, see Table 1 | ||||||||
Men | 190 (68) | 97 (65) | 93 (72) | 0.22 | 37 (76) | 15 (68) | 38 (68) | 0.57 |
Age at baseline, y | 39 (13) | 40 (13) | 38 (13) | 0.16 | 41 (13) | 41 (10) | 34 (12) | 0.009 |
Age at diagnosis, y | 35 (13) | 36 (14) | 34 (13) | 0.17 | 37 (12) | 37 (12) | 30 (13) | 0.02 |
Acute onset | 54 (19) | 28 (19) | 26 (20) | 0.78 | 13 (27) | 1 (5) | 11 (20) | 0.19 |
Familial DCM | 152 (54) | 54 (36) | 98 (75) | <0.001 | 39 (80) | 20 (91) | 37 (66) | <0.001 |
Family history of SCD <50 y | 20 (7) | 8 (5) | 12 (9) | 0.21 | 2 (4) | 5 (23) | 5 (9) | 0.04 |
SCA | 20 (7) | 9 (6) | 11 (8) | 0.43 | 1 (2) | 4 (18) | 6 (11) | 0.07 |
ICD as secondary ppx | 23 (8) | 10 (7) | 13 (10) | 0.31 | 1 (2) | 6 (27) | 6 (11) | 0.006 |
ICD as primary ppx | 47 (17) | 23 (15) | 24 (18) | 0.49 | 12 (24) | 4 (18) | 6 (11) | 0.28 |
CRT | 18 (6) | 12 (8) | 6 (5) | 0.25 | 2 (4) | 3 (14) | 1 (2) | 0.16 |
PM for bradyarrhythmia | 35 (13) | 15 (10) | 20 (15) | 0.17 | 4 (8) | 13(59) | 2 (4) | <0.001 |
Atrial arrhythmia, n = 271 | 75 (28) | 31 (21) | 44 (35) | 0.01 | 17 (35) | 14 (64) | 12 (23) | <0.001 |
AV block, n = 266 | 56 (21) | 22 (15) | 34 (28) | 0.01 | 9 (18) | 19 (86) | 5 (10) | <0.001 |
AV block >1°, n = 266 | 26 (10) | 12 (8) | 14 (11) | 0.41 | 2 (4) | 12 (55) | 0 | <0.001 |
LBBB, n = 266 | 70 (26) | 49 (34) | 21 (17) | 0.002 | 8 (16) | 9 (50) | 3 (6) | <0.001 |
HF | 232 (83) | 128 (85) | 104 (80) | 0.24 | 43 (88) | 17 (77) | 41 (73) | 0.13 |
NYHA class 3–4 | 58 (21) | 31 (21) | 27 (21) | 0.99 | 10 (20) | 6 (27) | 8 (15) | 0.65 |
LVDD, mm, n = 275 | 64 (10) | 65 (10) | 64 (10) | 0.28 | 65 (9) | 59 (9) | 64 (10) | 0.054 |
LVEF, %, n = 275 | 35 (12) | 34 (12) | 35 (12) | 0.6 | 35 (11) | 37 (13) | 36 (13) | 0.7 |
LVEF <30%, n = 275 | 87 (32) | 46 (31) | 41 (32) | 0.83 | 16 (33) | 6 (27) | 16 (30) | 0.97 |
RV dysfunction, n = 275 | 62 (23) | 33 (22) | 29 (23) | 0.92 | 13 (27) | 5 (23) | 9 (17) | 0.71 |
Comorbidities | ||||||||
Muscular dystrophy | 9 (3) | 3 (2) | 6 (5) | 0.31 | 0 | 1 (5) | 5 (9) | 0.04 |
CK activity >350 IU/l, n = 248 | 15 (6) | 8 (6) | 7 (6) | 0.96 | 1 (2) | 1 (5) | 5 (11) | 0.42 |
CAD | 7 (3) | 3 (2) | 4 (3) | 0.71 | 3 (6) | 1 (5) | 0 | 0.14 |
AH | 51 (18) | 34 (23) | 17 (13) | 0.04 | 12 (24) | 0 | 4 (7) | 0.005 |
Diabetes | 16 (6) | 12 (8) | 4 (3) | 0.08 | 1 (2) | 0 | 2 (4) | 0.32 |
Medication | ||||||||
β-Blocker | 235 (84) | 129 (86) | 106 (82) | 0.31 | 42 (86) | 18 (82) | 45 (80) | 0.76 |
ACEI/ARB/ARNI | 239 (85) | 130 (87) | 109 (84) | 0.51 | 45 (92) | 20 (91) | 43 (77) | 0.12 |
MRA | 146 (52) | 81 (54) | 65 (50) | 0.50 | 33 (67) | 7 (32) | 24 (43) | 0.02 |
Diuretics | 122 (44) | 67 (45) | 55 (42) | 0.69 | 32 (65) | 7 (32) | 15 (27) | <0.001 |
Therapy for acute HF | 18 (6) | 10 (7) | 8 (6) | 0.86 | 2 (4) | 0 | 4 (7) | 0.73 |
At the time of baseline evaluation, 83% of our patients were diagnosed with HF, 21% were in the NYHA class 3 or 4. The mean (SD) baseline LVEF was 35% (12%), and 31% had LVEF below 30%. One‑fourth of the participants had ICD implanted (33% as secondary prophylaxis), and 6% received CRT.
In the entire cohort, 3% of the patients were diagnosed with muscular dystrophy, and 6% had elevated activity of serum creatine kinase at above 350 IU/l. Comorbidities were infrequent, with arterial hypertension (AH) being the most common, found in 18% of the patients.
Atrial arrhythmias and AVB were more frequent in the gene‑positive DCM (34% vs 21%; P = 0.009 and 26% vs 15%; P = 0.01, respectively). In contrast, left bundle branch block (LBBB) and AH were more common in the gene‑negative group (16% vs 34%; P = 0.001 and 13% vs 23%; P = 0.03, respectively).
The patients from the group 3 (non-TTN, non-LMNA variant carriers) were characterized by younger age at diagnosis and baseline evaluation. LMNA-variant carriers had more adverse family history, and were more likely to have arrhythmias and conduction disorders, including those requiring device implantation. AH was more common in the gene‑negative DCM and in TTN-variant carriers.
Clinical course and prognosis
Median (IQR) follow‑up was 6.2 (2.7–8.9) years.
In the study cohort, 49 patients underwent HTx and 35 died (deaths after HTx not included). Of the deaths, 34 were cardiovascular, primarily due to progressive HF; 4 were sudden. LVAD was used in 25 patients: in 17 as a bridge to HTx, and in 3 patients as long‑term treatment. Two patients died shortly after implantation from complications, and in another 3 patients, the device was explanted after improvement.
ICD was implanted in 145 patients (70 before initial assessment, the rest during follow‑up). In secondary prevention, ICD was implanted in 25 patients: in 19 after SCA (in 11 patients it was the first symptom of the disease) and in 6 following a diagnosis of sustained, hemodynamically unstable VT. CRT was used in 55 patients, in most of them (n = 52) together with a defibrillation system, and in 3 patients with pacing only.
A total of 177 participants developed severe DCM, and 89 reached the composite end point of cardiovascular death, HTx, or LVAD implantation.
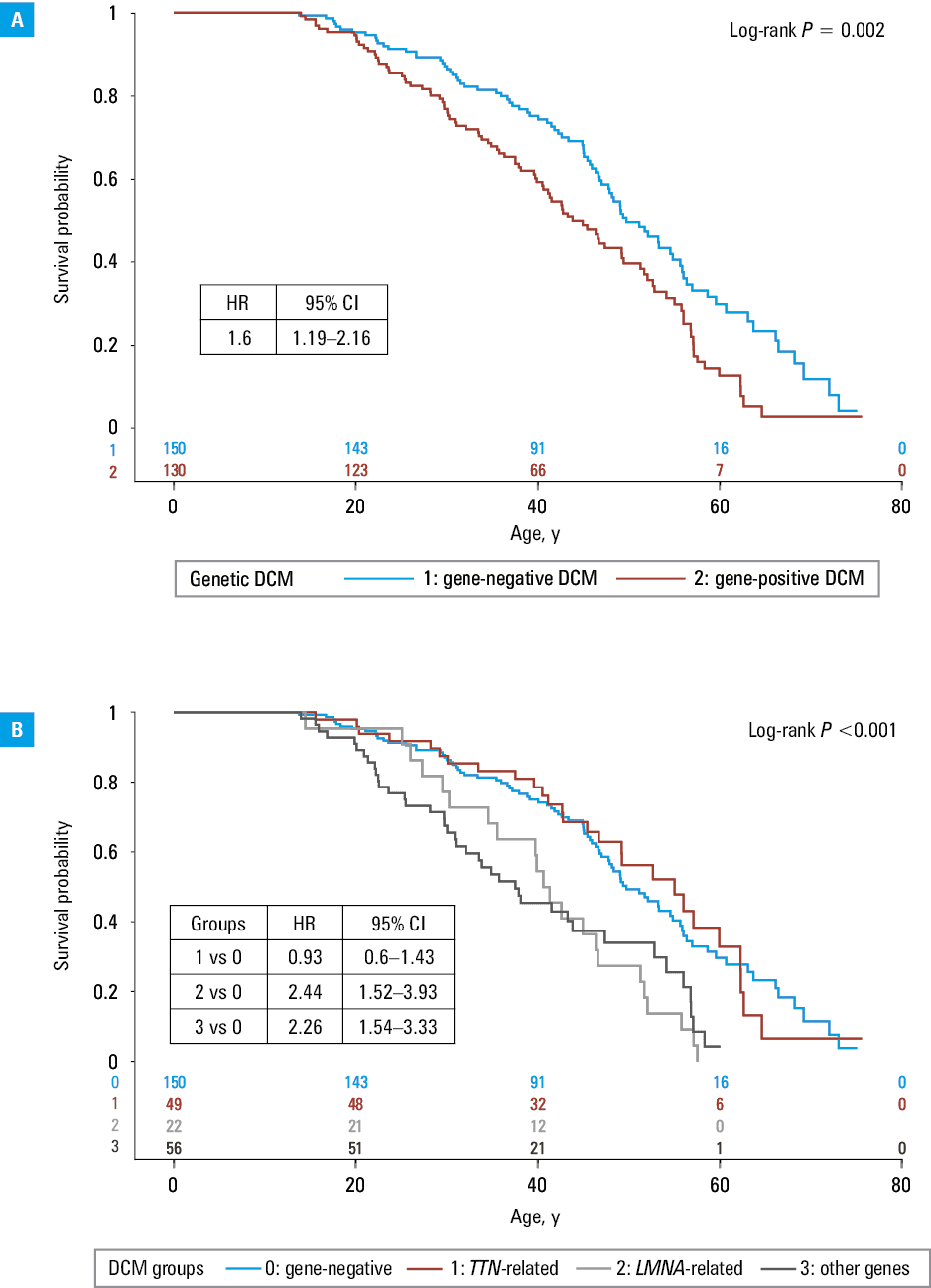
The gene‑positive group had worse prognosis, both in terms of the risk of developing severe DCM (Figure 2A; HR, 1.6; 95% CI, 1.19–2.16) and a composite end point of cardiovascular death, HTx, or LVAD implantation (HR, 1.81; 95% CI, 1.19–2.77; Figure 3A), but it varied depending on the genetic background.
Abbreviations: HR, hazard ratio; others, see Table 1
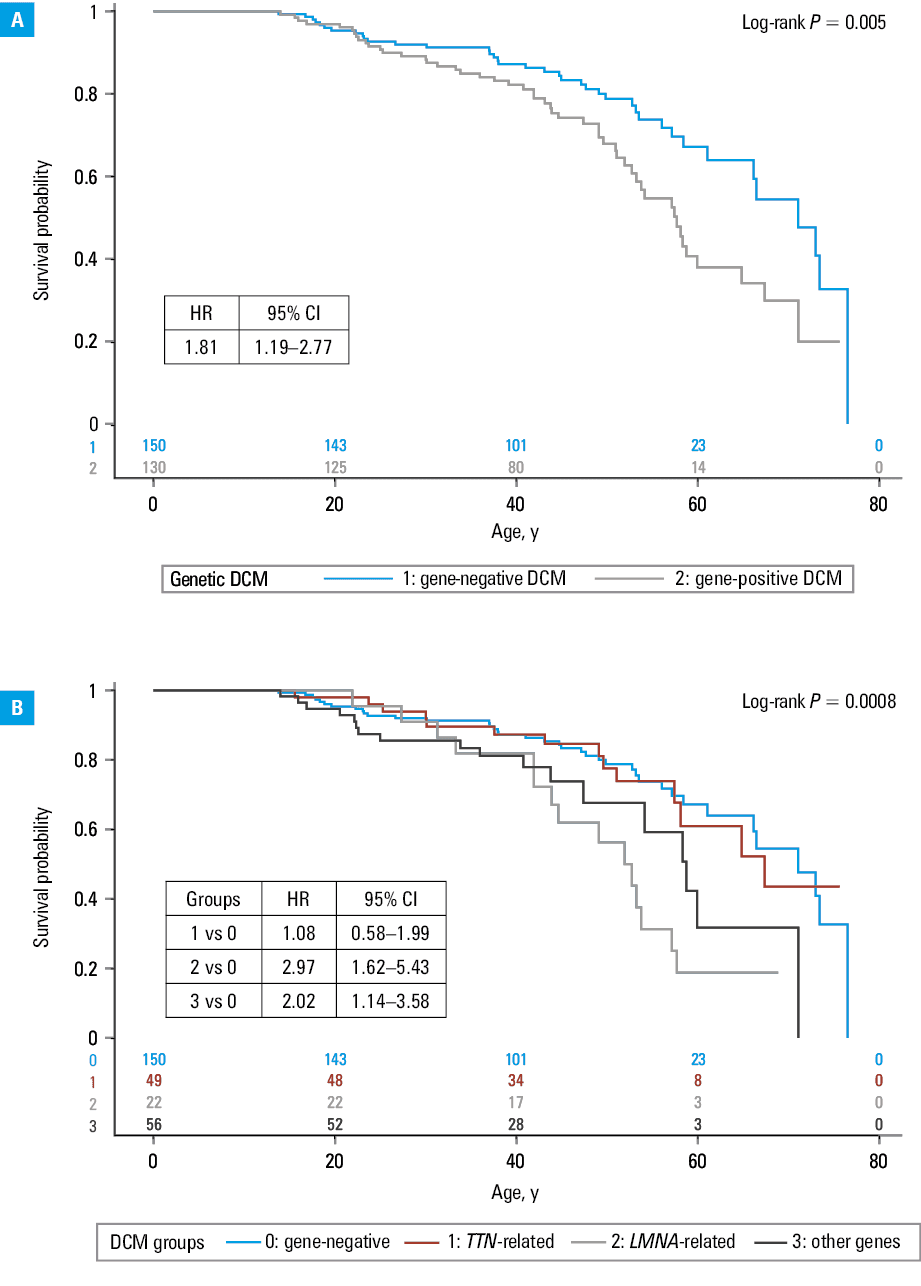
The prognosis in the TTN variant carriers did not differ from the gene‑negative group (HR, 0.93; 95% CI, 0.6–1.43 for developing severe DCM and HR, 1.08; 95% CI, 0.58–1.99 for cardiovascular death, HTx, or LVAD implantation).
In contrast, the risk of developing severe DCM was higher in the carriers of P/LP variants in the LMNA or other genes (Figure 2B). In comparison with gene‑negative DCM, it was 2.4‑fold higher in the group 2 (LMNA-variant carriers), and 2.3‑fold higher in the group 3 (95% CI, 1.52–3.93 and 1.54–3.33, respectively). Also, the risk of cardiovascular death, HTx, or LVAD implantation was higher in the groups 2 and 3 than in the gene‑negative group (HR, 2.97; 95% CI, 1.62–5.43 and HR, 2.02; 95% CI, 1.14–3.58, respectively; Figure 3B).
Discussion
In our single‑center study of carefully phenotyped Polish patients with DCM, clinically actionable genetic variants could be identified in as many as 46% of participants. Clinical importance refers primarily to variants associated with a high risk of SCD regardless of LVEF, particularly to LMNA, filamin (FLNC), DSP, RNA‑binding motif protein 20 (RBM20), and PLN variants.1 Also, identifying the carriers of SCN5A variants is crucial for tailoring optimal therapy, as they may respond favorably to sodium channel blockers.27-29 Equally important is to identify those relatives of DCM patients who are carriers of a causative variant, and are at a risk of developing the disease.1,30,31
The yield of genetic testing in our study is higher than in other studies. However, comparisons are difficult and several caveats must be taken into account: 1) differences in the set of genes assessed, different in each study; it should be emphasized that the association of the FLNC gene with DCM was found recently and only since 2018 has this gene been widely included in genetic panels dedicated to DCM; 2) differences in the assessment of variant pathogenicity, especially before the adoption of the ACMG criteria,21 but also later, because the pathogenicity assessment according to the ACMG criteria has also been evolving22,30; 3) the effectiveness of genetic testing may vary depending on the characteristics of the study group, for example, age, disease stage, incidence of the familial disease, or a burden of comorbidities.9,12
In a recent report from southeastern Poland,16 the yield of genetic testing was lower and amounted to 24%, even though the authors included genes of limited association with DCM (eg, MYBPC3, MYPN) and variants of uncertain pathogenicity (eg, missense variants in TTN and FLNC). The study group, in comparison with ours, was older (mean age, 45 vs 39 y) and had a lower mean LVEF (30% vs 35%).
A high yield of positive genetic results (37%) was obtained both in the Spanish DCM cohort reported by Escobar‑Lopez et al10 and in the Italian‑American cohort of Familial Cardiomyopathy Registry reported by Gigli et al.11 Both cohorts were characterized by a high frequency of familial cases (48% and 60%, respectively) and advanced LV systolic dysfunction (mean LVEF, 32% and 33%, respectively), with a mean age of 53 and 41 years, respectively. The effectiveness of genetic testing was lower in the Dutch Maastricht Cardiomyopathy Registry (18%) and in the US multisite DCM Precision Medicine Study (19%).9,12,30 Of note, in the latter study, a large discrepancy between individuals of the European and African ancestry was noted (26% vs 8%).9 The study cohort was characterized by advanced HF (mean LVEF, 21%) but also a high frequency of comorbidities (AH, 53% and diabetes, 25% vs 18% and 6%, respectively, in our group), especially in the individuals of African ancestry (68% and 31%, respectively).9,12 P/LP variants were detected more frequently in the group that required HTx or LVAD implantation.12
The above comparisons indicate factors that may have influenced the unusually high yield of genetic testing in our cohort. Many DCM patients referred to our tertiary center from all over Poland were young but with advanced HF, often candidates for HTx, with a low burden of comorbidities, but often with a positive family history. At the same time, these factors limit the extrapolation of our results to the general DCM population. It is also of some importance that among the genes studied, we included DMD, which is markedly associated with DCM, but was not included by the ClinGen experts or in some of the cited studies. On the other hand, in our cohort, FLNC was not tested in 40% of the patients, including 19% (n = 52) in whom no other genetic cause of DCM was detected.
In our study, the distribution of gene variants was generally similar to other studies, with TTN and LMNA variants being the most frequent, however, the values varied between studies. TTN variants accounted for 38% of gene‑positive DCM in our study and in that by Escobar Lopez et al,10 for 30% of gene‑positive DCM in the study by Gigli et al,11 and as much as 60% in the study by Hofmeyer et al.12 In the case of LMNA variants, the frequency was 9%–10% of gene‑positive DCM in the abovementioned studies, and almost twice as high in our study (17%). We identified only 1 pathogenic RBM20 variant. RBM20-related DCM is known to be associated with poor prognosis, especially in men,32 and variants in this gene were found more frequently in other populations (2.2%–5.4% of gene‑positive DCM).10-12 We did not identify any founder mutations in the Polish population, unlike in the Netherlands, where the PLN Arg14del variant is prevalent in patients with DCM.
It is worth noting, however, that we detected the TTN c.82432del variant in as many as 4 participants and the LMNA c.575A>G variant in 3 individuals.
We found a poorer prognosis among the patients with gene‑positive DCM, and more precisely in those carrying P/LP variants in the genes other than TTN. The prognosis in TTN-related DCM did not differ from that of gene‑negative DCM. These results are consistent with the observations made in the Spanish and Dutch cohorts. In the study by Escobar‑Lopez et al,10 HR of major adverse cardiovascular events over a 4‑year follow‑up period was 1.5. Similarly, Stroeks et al30 showed that patients with a P/LP gene variant had shorter event‑free survival than those with gene‑negative DCM, regardless of the number of genes in the used panels. Also, a trend toward more frequent malignant arrhythmia (P = 0.06) and end‑stage HF (P = 0.06) was found in gene‑positive DCM over a median follow‑up of 10 years by Gigli et al,11 although they found no difference in all‑cause mortality. It should be noted that the end points used in each study differed. Furthermore, unlike in other cited studies, we decided to assess survival from birth to show the prognosis dependent on the genetic background throughout life, not at the time of the disease diagnosis.
Limitations
A major limitation of the study is that it was conducted in a single tertiary referral center, and therefore it may have included more young patients, those with poorer prognosis and a burdensome family history in comparison with the general DCM population. Moreover, although the patients included came from all over Poland, the geographic distribution of their places of residence was not even. The patients living closer to the center predominated, while those from distant regions were less numerous.
Conclusions
In summary, DCM‑related genetic variants were identified in almost half of the patients from our study cohort. The patients with identified variants were characterized by a higher frequency of a positive family history, AVB or atrial arrhythmias, and a lower frequency of LBBB or AH. The prognosis was worse in the patients with variants in genes other than TTN.
- Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023; 44: 3503‑3626. | Crossref
- Schultheiss HP, Fairweather D, Caforio ALP, et al. Dilated cardiomyopathy. Nat Rev Dis Primers. 2019; 5: 32. | Crossref
- Hershberger RE, Cowan J, Jordan E, et al. The complex and diverse genetic architecture of dilated cardiomyopathy. Circ Res. 2021; 128: 1514‑1532. | Crossref
- Haas J, Frese KS, Peil B, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015; 36: 1123‑1135a. | Crossref
- Garnier S, Harakalova M, Weiss S, et al. Genome‑wide association analysis in dilated cardiomyopathy reveals two new players in systolic heart failure on chromosomes 3p25.1 and 22q11.23. Eur Heart J. 2021; 42: 2000‑2011. | Crossref
SUPPLEMENTARY MATERIAL
ARTICLE INFORMATION