Primary extraosseous Ewing sarcoma / primitive neuroectodermal tumor of head and neck in an adult

 CC BY 4.0
CC BY 4.0
Primary extraosseous Ewing sarcoma / primitive neuroectodermal tumor of head and neck in an adult
A 56‑year‑old man was admitted to our department due to rapid tumor growth on the right cheek, without signs of difficulty swallowing, breathing, or visual disturbances. His medical history included stenting of the coronary artery and antihypertensive treatment, with a family history negative for oncological diseases. He was a heavy smoker. On physical examination, partial skin necrosis over the tumor was observed, along with enlarged, hard submandibular lymph nodes on the right side, moderate trismus, and a 4 cm × 3 cm intraoral ulceration of the cheek (Figure 1A). One month prior, the patient presented only with mild swelling. Radiologic examinations excluded dental causes, facial bone fractures, or inflammation. Based on his history of facial trauma and the presence of purulent contents on aspiration biopsy, a hematoma progressing to abscess was initially suspected. The abscess was drained, and empirical antimicrobial therapy was administered. The patient did not attend follow‑up for a month.
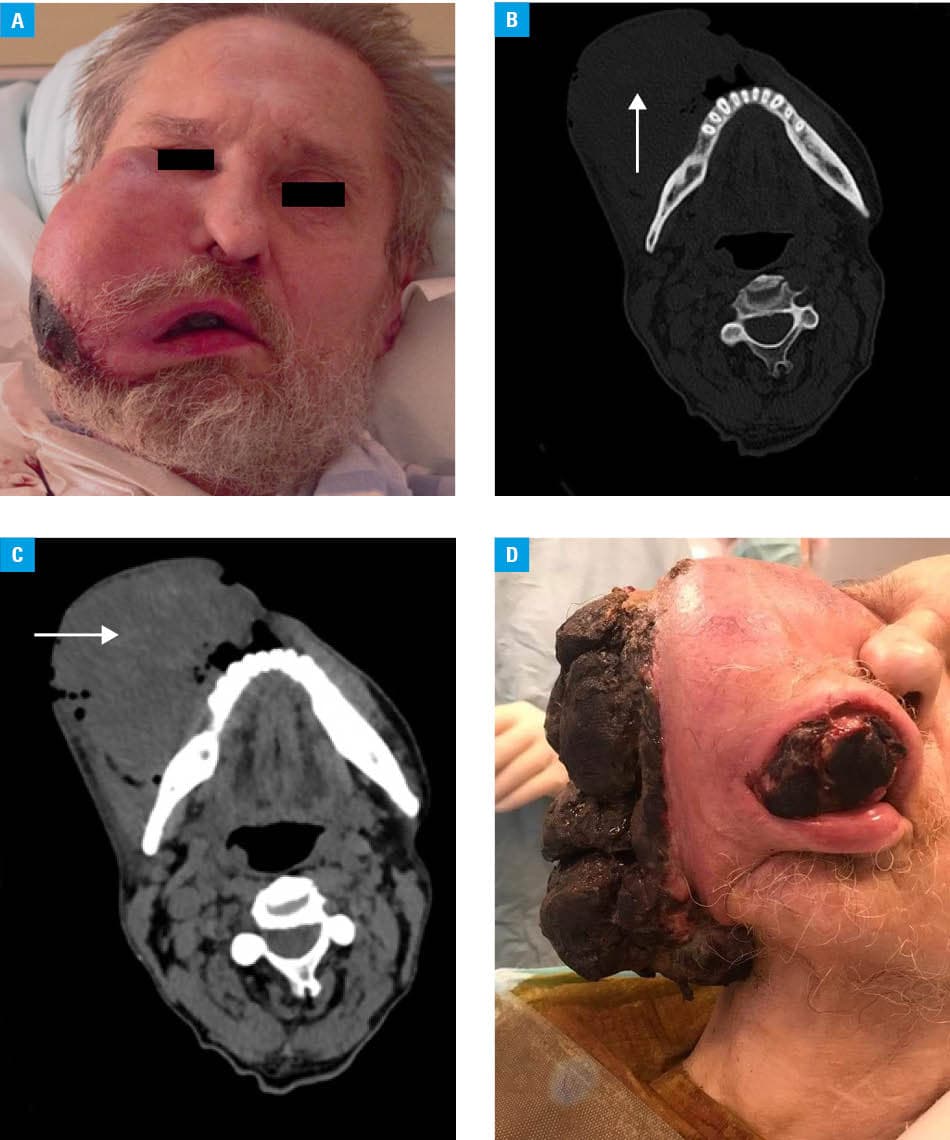
Upon admission, contrast‑enhanced computed tomography (CT) of the head and neck showed an extensive, heterogeneously enhancing tumor with central necrosis, reaching the lateral angle of the eye in the upper pole and the submental region in the lower pole, measuring 65 mm × 95 mm × 115 mm (Figure 1B). No osteolysis was detected. Whole‑body CT was performed to exclude dissemination of the primary disease. Laboratory tests showed an elevated C‑reactive protein level (243.56 mg/l; reference range <5 mg/l). Excisional biopsy was performed; however, while waiting for histopathology results, the tumor grew rapidly (Figure 1C and 1D). Histopathology demonstrated Ewing sarcoma / primitive neuroectodermal tumor (ES/PNET), with the phenotype: CD99+, CK–, LCA–, SMA–, CD117–, CD34+, CD33–, S‑100–, synaptophysin–, melanA–, desmin–, CD56–, CD57–, and PAS–.
Given the diagnosis, the patient received 1 cycle of chemotherapy in the vincristine, doxorubicin, and cyclophosphamide (VDC) regimen at 100 mg, 60 mg, and 2 mg, respectively, which was moderately well tolerated. Further chemotherapy was discontinued due to the patient’s anemia and tumor progression with persistent ulcer bleeding. The patient was qualified for palliative hemostatic radiotherapy at a dose of 20 Gy (5 fractions of 4 Gy), achieving hemostasis in the tumor. However, systemic treatment was halted due to his poor general condition, and palliative care measures were implemented. In the subsequent months, the patient’s airways were secured with a tracheostomy, and percutaneous endoscopic gastrostomy was performed to maintain nutritional access. The patient died a few months later.
ES/PNET is an aggressive malignant tumor that can occur intraosseously or extracorporally. Extraskeletal ES/PNET is rare and usually affects the trunk and extremities. It commonly presents in children and young adults. Peripheral PNETs, arising outside the central nervous system (CNS), are thought to originate from migrating embryonal neural crest cells. Rare in the head and neck regions, few cases involve the maxilla, nasal cavity, maxillary sinus, or ethmoid sinus.1 ES typically affects individuals aged 11–20 years, with a predominance in men and higher incidence in white people. Head and neck ES accounts for only 3%–9% of the cases, often presenting as a rapidly enlarging, painful mass with possible early CNS involvement.2-4
Histologically, ES appears as a small, blue, round cell tumor. Nearly 95% of the cases harbor a characteristic translocation between chromosomes 11q and 22q, which aids diagnosis. Due to its primitive appearance, ES/PNET can be challenging to diagnose; in differential diagnosis, it is important to consider lymphoma, rhabdomyosarcoma, poorly differentiated carcinoma, and Merkel cell carcinoma. Therefore, immunohistochemistry, cytogenetics, reverse transcription polymerase chain reaction, and fluorescence in situ hybridization are crucial for differentiating ES/PNET from other small round cell tumors.
Due to its rarity and aggressive nature, early diagnosis and a multidisciplinary approach are vital to improve prognosis, especially for head and neck presentations. Most current treatment protocols are based on combinations of the most active chemotherapeutic agents, including doxorubicin, cyclophosphamide, vincristine, etoposide, ifosfamide, and dactinomycin. The most frequently proposed regimen is an interval‑compressed protocol with 2‑week intervals, utilizing VDC, alternating with ifosfamide and etoposide. Recommended local treatments include wide‑margin en‑bloc resection with at least 1 cm of healthy tissue and definitive radiotherapy using doses in the range of 54–60 Gy.5
- Kulkarni MM, Khandeparkar SGS, Joshi AR, Barpande C. A rare case of extraskeletal Ewing’s sarcoma / primitive neuroectodermal tumor developing in maxillary sinus of an old patient. J Oral Maxillofac Pathol. 2016; 20: 330 | Crossref
- Ellis MA, Gerry DR, Neskey DM, Lentsch EJ. Ewing sarcoma of the head and neck. Ann Otol Rhinol Laryngol. 2017; 126: 179‑184. | Crossref
- Grevener K, Haveman LM, Ranft A, et al. Management and outcome of Ewing sarcoma of the head and neck. Pediatr Blood Cancer. 2016; 63: 604‑610. | Crossref
- Chin EW, Abu‑Bakar AZ, Hitam S, et al. Primary extraosseous Ewing sarcoma of the maxillary sinus in an adult‑a rare case report. Iran J Otorhinolaryngol. 2019; 31: 391‑397. | Crossref
- Mata Fernández C, Sebio A, Orcajo Rincón J, et al. Clinical practice guidelines for the treatment of Ewing sarcoma (Spanish Sarcoma Research Group‑GEIS). Clin Transl Oncol. 2025; 27: 824‑836. | Crossref
ARTICLE INFORMATION