First-in-class oral hypoxia-inducible factor 2α inhibitor in von Hippel–Lindau disease: a new era of targeted therapy
 ,
,

 CC BY 4.0
CC BY 4.0
First-in-class oral hypoxia-inducible factor 2α inhibitor in von Hippel–Lindau disease: a new era of targeted therapy
A 58‑year‑old man with genetically confirmed von Hippel–Lindau (VHL) syndrome (estimated prevalence of 1 per 36 000 population; approximately 900 genetically confirmed patients in Poland) presented with a complex, multisystem disease course.1 His history included left nephrectomy for clear cell renal carcinoma (1995) and partial right nephrectomy for multifocal lesions (2005). Between 2013 and 2017, he underwent multiple neurosurgical procedures for recurrent hemangioblastomas of the cerebellum and spinal cord. In December 2022, he required radicalization of the right nephrectomy, and has since been on hemodialysis. In 2020 and 2021, additional stereotactic radiosurgery (Gamma Knife; Elekta, Stockholm, Sweden) was performed for cerebellar hemangioblastomas.
Further manifestations included pancreatic cysts and initial suspicion of left adrenal pheochromocytoma, as well as a pancreatic neuroendocrine tumor. However, these lesions were not confirmed on imaging performed at our center, nor on somatostatin receptor scintigraphy or gallium‑68 positron emission tomography / computed tomography. The presence of pheochromocytoma was also not supported by plasma assays of methoxy‑catecholamines performed with liquid chromatography–mass spectrometry. Slightly elevated plasma metanephrine levels were attributed to chronic kidney disease rather than true catecholamine excess.
At the beginning of October 2024, belzutifan therapy was initiated through a Named Patient Program, as the drug had not yet been authorized in Europe. The treatment was consistent with the Polish Ministry of Health regulations and supported by the 2023 recommendation of the Polish Agency for Health Technology Assessment and Tariff System2 on the use of belzutifan in VHL syndrome. The decision was made because of progression of neurologic symptoms, and a high risk associated with further neurosurgical interventions. Belzutifan selectively inhibits the hypoxia‑inducible factor 2α (HIF‑2α) pathway, thereby blocking downstream angiogenesis and tumor growth.3,4 The patient reported considerable improvement in neurological function, including reduced vertigo, improved balance, normalization of the finger‑to‑nose and Romberg tests, and improved hearing in the left ear after 3–4 months of therapy. The only significant adverse effect was moderate anemia (hemoglobin, 9.6 g/dl; reference range, 13–18 g/dl), a predictable on‑target effect of HIF‑2α inhibition.5 Brief erythropoietin (EPO) therapy during hemodialysis was discontinued after hemoglobin stabilization. The patient has since remained stable without EPO use or transfusions, and no tumor progression was observed on follow‑up examination.
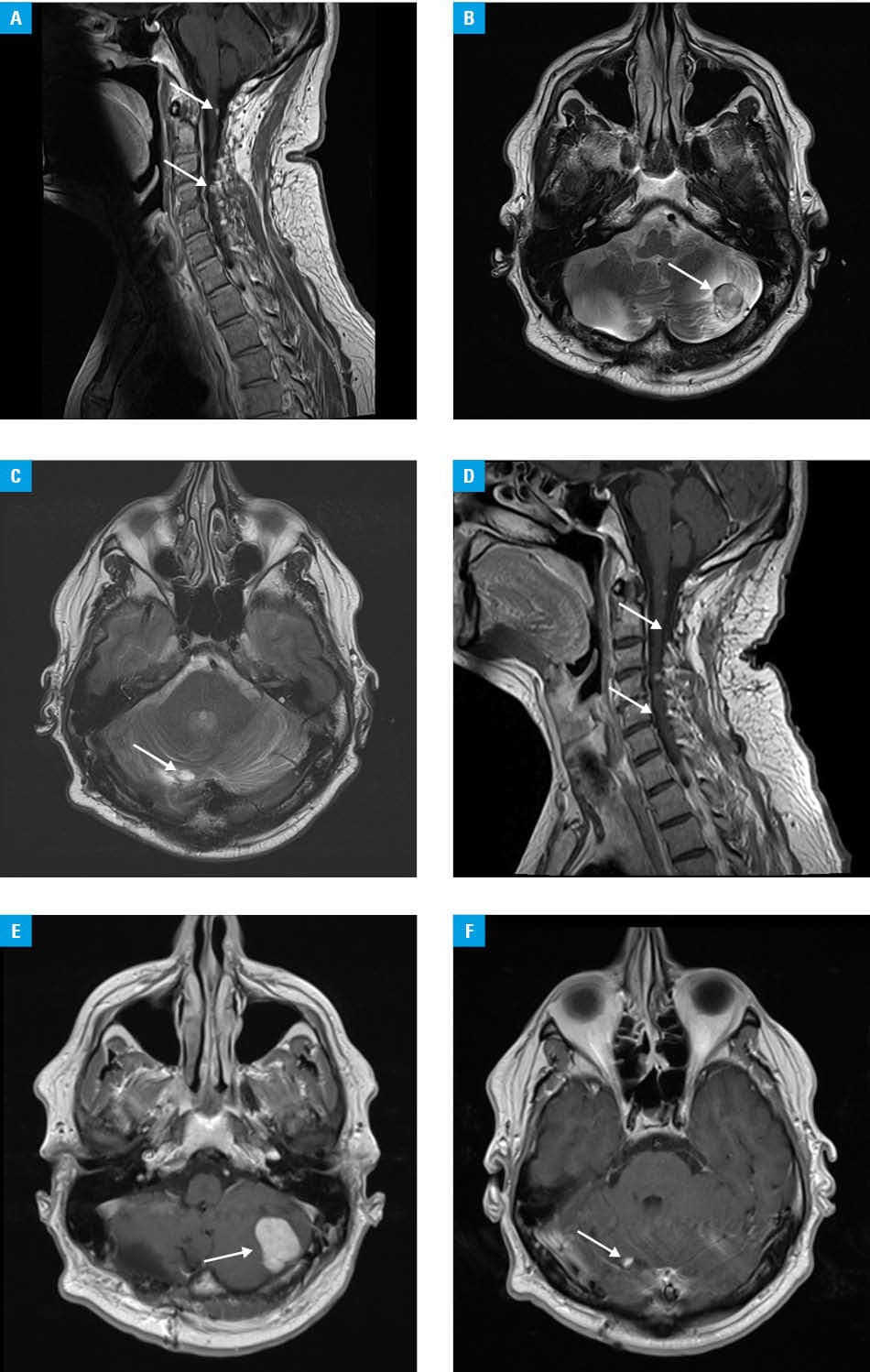
Follow‑up magnetic resonance imaging (October 2024 vs January 2025; Figure 1A–1C vs 1D–1F) showed a regression of most hemangioblastomas in the spinal cord and cerebellum. The largest cerebellar lesion displayed slight enlargement, but with marked reduction of peritumoral edema, correlating with clinical improvement (Figure 1B vs 1E). These findings are consistent with the LITESPARK‑004 trial,5 which reported an objective response rate of approximately 44% in VHL‑associated hemangioblastomas. Anemia is a predictable on‑target effect resulting from suppressed endogenous EPO production, and may serve as a biomarker of effective HIF‑2α pathway inhibition. This case illustrates the efficacy and tolerability of belzutifan, the first molecularly‑targeted therapy for VHL syndrome. The treatment led to radiologic regression of most central nervous system hemangioblastomas, reduction of peritumoral edema, and neurologic improvement. Belzutifan shows how targeted oral therapy can improve prognosis and quality of life in patients with this rare multisystem disease.
- VHL Alliance. Epidemiology and clinical manifestations of von Hippel–Lindau disease. https://vhl.org. Accessed September 15, 2025.
- Welireg (belzutifan) in von Hippel–Lindau disease: assessment report. Warsaw: Polish Agency for Health Technology Assessment and Tariffication (AOTMiT); 2023. https://bip.aotm.gov.pl/assets/files/zlecenia_mz/2023/006/RPT/6_OT.4211.3.2023_Welireg_[belzutifan]_VHL_BIP.pdf. Accessed September 25, 2025.
- Jonasch E, Donskov F, Iliopoulos O, et al. Belzutifan for renal cell carcinoma in von Hippel–Lindau disease. N Engl J Med. 2021; 385: 2036‑2046. | Crossref
- Choueiri TK, Kaelin WG Jr. Targeting the HIF2‑VEGF axis in renal cell carcinoma. Nat Med. 2020; 26: 1519‑1530. | Crossref
- Iliopoulos O, Iversen AB, Narayan V, et al. Belzutifan for patients with von Hippel–Lindau disease‑associated CNS haemangioblastomas (LITESPARK‑004): a multicentre, single‑arm, phase 2 study. Lancet Oncol 2024; 25: 1325‑1336. | Crossref
ARTICLE INFORMATION