Newly diagnosed homocystinuria as a rare cause of advanced peripheral artery disease in a 41-year-old man

 CC BY 4.0
CC BY 4.0
Newly diagnosed homocystinuria as a rare cause of advanced peripheral artery disease in a 41-year-old man
Homocystinuria is a rare (1 per 200 000 to 335 000 individuals)1 inborn metabolic disorder with a median (interquartile range) age at diagnosis of 10 (0.1–73) years,2 caused by missense mutations in the cystathionine β-synthase (CBS) gene, resulting in high total homocysteine (tHcy) and methionine concentrations in the blood and tissues. Clinical manifestations of homocystinuria in adults include skeletal symptoms, ectopia lentis, a Marfan syndrome‑like appearance, premature atherosclerosis, and thromboembolic events.1,3 Homocystinuria may cause early‑onset peripheral artery disease (PAD), and here, we present such a case.
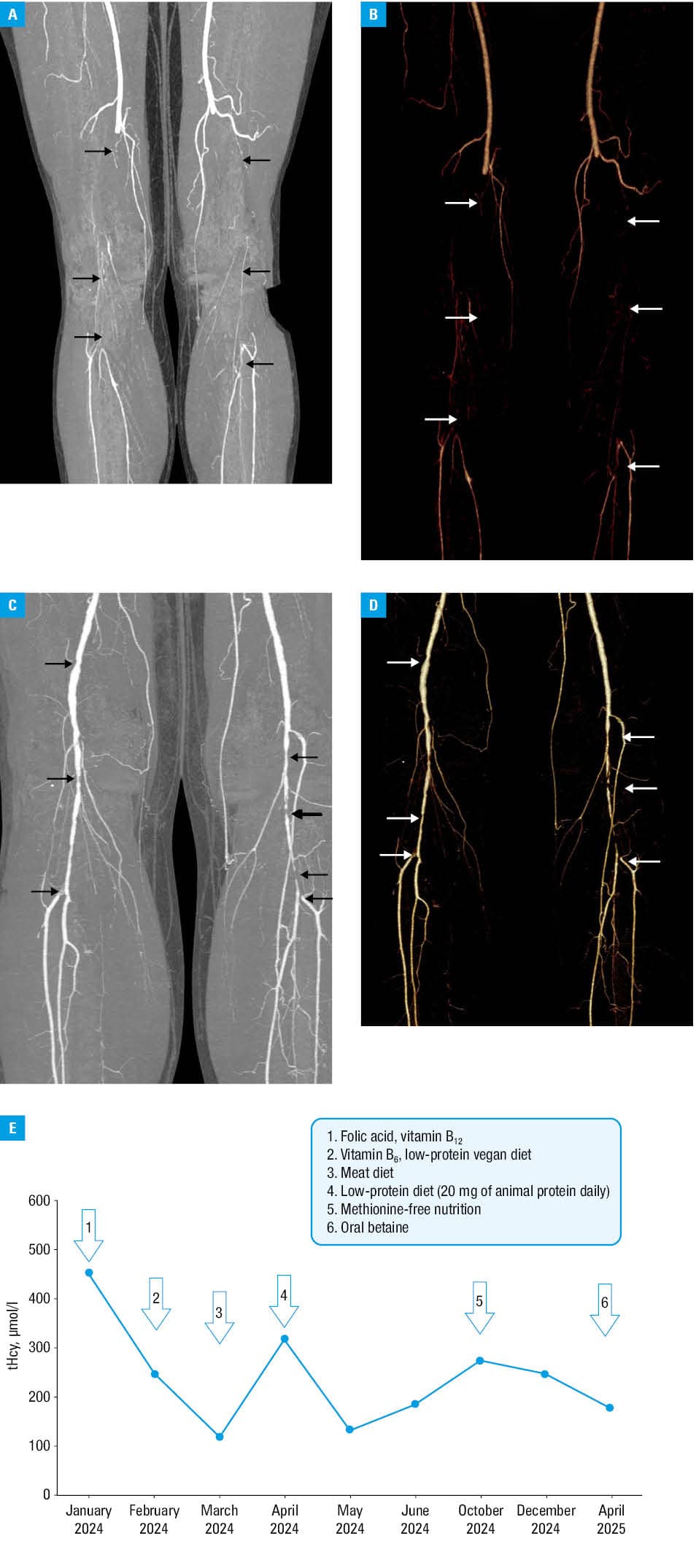
A 38‑year‑old man, previously asymptomatic, nonsmoker, with obesity (height, 202 cm; weight, 126 kg; body mass index [BMI], 30.9 kg/m2), hypertension, and hypercholesterolemia (low‑density lipoprotein cholesterol [LDL‑C], 3.6 mmol/l; reference range [RR] <3 mmol/l) experienced paroxysmal atrial fibrillation. His family history for cardiovascular disease was unremarkable. Aspirin (75 mg/d), rivaroxaban (20 mg/d), atorvastatin (20 mg/d), ezetimibe (10 mg/d), and antihypertensive therapy were initiated. In March 2022, he experienced acute ischemia of the lower extremities. Computed tomography angiography showed critical stenoses of the superficial femoral arteries with distal acute thrombosis (Figure 1A and 1B), which was successfully treated with thrombolysis and mechanical thrombectomy of the left popliteal artery. In October 2022, the patient suffered acute thromboembolism of the left popliteal and tibial arteries (Figure 1C and 1D). Attempts at thrombectomy and angioplasty failed, and due to ongoing ischemia and septic shock, the left lower extremity was amputated below the knee.
Routine thrombophilia screening showed no abnormalities. Lipoprotein(a) level was low.
In July 2023, the patient was diagnosed with bilateral lens luxation, left retinal detachment with subsequent lens implantation in the right eye and left eye vitrectomy; however, he lost sight in the left eye.
In early 2024, the patient was referred to our center with suspicion of atypical thrombophilia. Further examinations showed extremely high fasting plasma tHcy concentration (454.76 µmol/l; RR <15 µmol/l) with common heterozygous variants of the 5,10‑methylenetetrahy‑drofolate reductase gene c.665C>T (p.Ala222Val) and c.1286A>C (p.Glu429Ala), which could not explain such tHcy levels. Next generation sequencing showed he was a carrier of compound heterozygous CBS variants c.833T>C (p.Ile278Thr) and c.1358+2T>A. The latter had once been reported to ClinVar database as “probably pathogenic,” associated with the classic form of homocystinuria, and it was predicted to disrupt RNA splicing by affecting the donor site in intron 14, which may lead to a loss of transcript in the nonsense‑mediated decay pathway.4 To our knowledge, this is the first report of a coexistence of these genetic variants. In the patient’s asymptomatic children, the presence of both CBS variants on separate alleles in a trans configuration was found: one daughter carried the c.833T>C (p.Ile278Thr) variant and the other one, the c.1358+2T>A variant; both had plasma tHcy levels below 15 µmol/l.
A pyridoxine challenge test confirmed B6-responsive homocystinuria. The patient was advised to follow a low‑protein vegan diet, reduce methionine intake, and take folic acid (40 mg/d), vitamin B12, (500 µg/d), vitamin B6 (25 mg/d) supplementation, which resulted in tHcy level falling to 100–200 µmol/l over 3 months. Due to the suboptimal tHcy level (>50 µmol/l; Figure 1E), oral betaine (6 g/d) was added.
As of July 2025, the patient was in a good condition, with BMI of 29.4 kg/m2, on apixaban (2 × 5 mg/d), along with other drugs, without thrombosis or PAD progression. The last LDL‑C level was 1.4 mmol/l, with some fluctuation due to nonadherence to pharmacotherapy. The patient is treated in a local outpatient cardiology clinic. The last echocardiography showed no abnormalities. Since the patient had no angina symptoms or signs suggestive of carotid artery stenosis, no imaging of coronary or carotid arteries has been performed so far.
This case underscores the importance of considering homocystinuria in a differential diagnosis of young adults with PAD, especially in the absence of strong cardiovascular risk factors. Apart from tHcy level drop, just as in the case of all other PAD patients,5 individuals with homocystinuria should achieve therapeutic goals recommended in hypercholesterolemia, hypertension, and obesity. tHcy measurement is inexpensive and widely accessible, and homocystinuria can be diagnosed and successfully managed in most countries, reducing the risk of severe complications. Due to residual atherosclerotic and thrombotic risk mainly associated with elevated tHcy, despite dietary intervention and vitamin‑based treatment, patients with homocystinuria require close surveillance by several specialists. Family counselling should be advised in every case.
- Bublil EM, Majtan T. Classical homocystinuria: from cystathionine β-synthase deficiency to novel enzyme therapies. Biochimie. 2020; 173: 48‑56. | Crossref
- Kožich V, Sokolová J, Morris AAM, et al. Cystathionine β-synthase deficiency in the E‑HOD registry‑part I: pyridoxine responsiveness as a determinant of biochemical and clinical phenotype at diagnosis. J Inherit Metab Dis. 2021; 44: 677‑692. | Crossref
- Morris AAM, Kožich V, Santra S, et al. Guidelines for the diagnosis and management of cystathionine β-synthase deficiency. J Inherit Metab Dis. 2017; 40: 49‑74. | Crossref
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17: 405‑424. | Crossref
- De Luca L. Who may benefit from low‑dose rivaroxaban plus aspirin? Practical implications for outpatients with cardiovascular disease. Pol Arch Intern Med. 2023; 133: 16566. | Crossref
ARTICLE INFORMATION