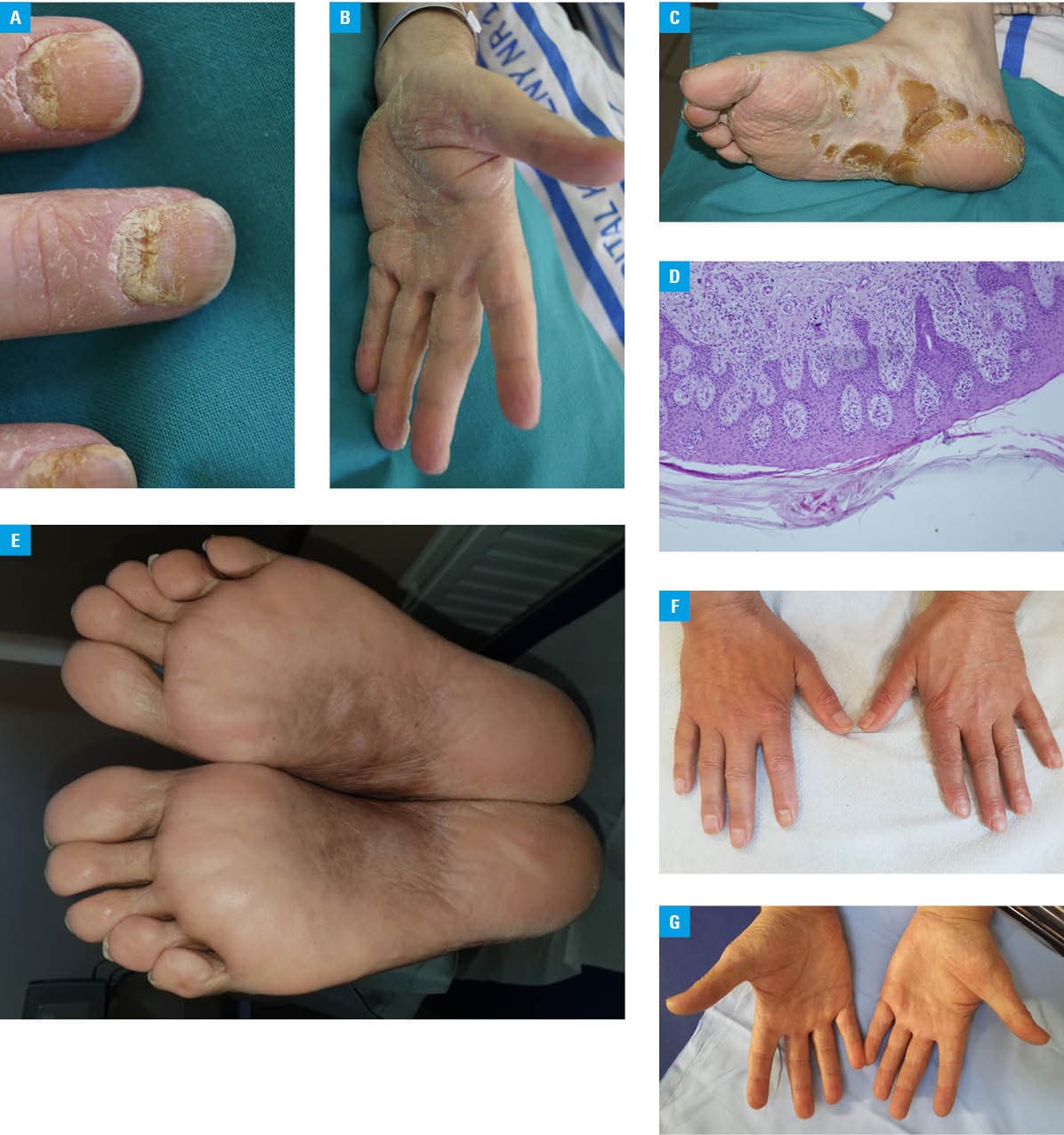
A 55‑year‑old man was referred to a hematology ward from a dermatology department for further diagnostic evaluation and treatment. The patient reported generalized weakness and bone pain localized to the spine and hip joints. On admission, he presented with hyperkeratotic lesions on the palms and soles, as well as hyperkeratosis of the fingernails. The lesions had first appeared 2 months prior to hospitalization and were resistant to standard topical therapy (Figure 1A–1C). The patient had no prior history of oncological treatment or dermatologic diseases, including psoriasis or eczema. Mycological examinations (direct microscopy and culture) of skin and nail scrapings were negative. Laboratory investigations showed normocytic anemia (hemoglobin, 8.5 g/dl; reference range [RR], 13–16.5 g/dl), elevated levels of serum creatinine (1.48 mg/dl; RR, 0.6–1.3 mg/dl) and C‑reactive protein (33.3 mg/l; RR, 0–5 mg/dl), markedly elevated free κ light chains (1390 mg/l; RR, 3.3–19.4 mg/l), and decreased free λ light chains (1.59 mg/l; RR, 5.71–26.3 mg/l). Serum immunofixation identified an immunoglobulin (Ig) G-κ monoclonal protein. Computed tomography visualized multiple osteolytic lesions with pathological fractures involving the Th5 and L3 vertebral bodies, as well as a fracture of the greater trochanter of the femur. Bone marrow aspiration showed 78% plasma cell infiltration, which was confirmed by immunophenotyping. Based on the clinical presentation and test results, a diagnosis of multiple myeloma, IgG-κ, stage IIIB was established. Histopathologic examination of skin biopsy specimens showed parakeratosis, hypergranulosis, and acanthosis (Figure 1D). The patient was treated with combination chemotherapy consisting of bortezomib, thalidomide, and dexamethasone (VTD regimen). After 3 months of therapy, a marked improvement in his general condition was noted, with significant regression of palmoplantar lesions. After 4 consecutive months of treatment, the cutaneous lesions completely resolved (Figure 1E–1G). Therefore, the patient was diagnosed with palmoplantar keratoderma (PPK) as a paraneoplastic manifestation of multiple myeloma (MM).
Cutaneous paraneoplastic syndromes represent a heterogeneous group of skin disorders associated with underlying malignancy.1 They are attributed either to tumor‑derived humoral mediators or to immune cross‑reactivity between neoplastic cells and normal skin.2 Their onset and clinical course parallel those of the primary malignancy.
PPK may be either acquired or inherited. Inherited forms of PPK are associated with autosomal dominant or autosomal recessive disorders. Mutations in several genes have been described in congenital PPK, including KRT1, KRT9, SLURP1, DSG1, KRT16, KRT6C, TRPV3, and POMP.3 Among their other effects, mutations in the genes encoding keratins or connexins lead to the formation of defective gap junctions.4 On the other hand, PPK can be classified as paraneoplastic syndrome, reported in association with various oncological diseases, most commonly gastrointestinal cancers (particularly esophageal and gastric carcinoma). Associations with urothelial carcinoma, pulmonary carcinoma, and lymphomas have also been documented.5-7 To our best knowledge, only a single case of PPK associated with MM has been previously described in the literature.8 The patient presented with both diffuse plane xanthomatosis and PPK, with marked improvement of cutaneous lesions following antimyeloma treatment. Similarly, in our case, resolution of palmoplantar keratoderma paralleled clinical improvement in the course of MM therapy. A diagnosis of paraneoplastic PPK should be considered in patients presenting with keratoderma refractory to standard dermatologic treatment, and appropriate malignancy screening is warranted. Management of PPK should primarily target the underlying malignancy, as treatment of the cancer typically results in regression of the skin manifestations. Cutaneous paraneoplastic syndromes are observed in a variety of hematologic malignancies, and in the cases of dermatologic disease with an unclear clinical course, a hematology consultation is recommended.
- Wick MR, Patterson JW. Cutaneous paraneoplastic syndromes. Semin Diagn Pathol. 2019; 36: 260‑271. | Crossref
- Pelosof LC, Gerber DE. Paraneoplastic syndromes: an approach to diagnosis and treatment. Mayo Clin Proc. 2010; 85: 838‑854. | Crossref
- Dev T, Mahajan VK, Sethuraman G. Hereditary palmoplantar keratoderma: a practical approach to the diagnosis. Indian Dermatol Online J. 2019; 10: 365‑379. | Crossref
- Avshalumova L, Fabrikant J, Koriakos A. Overview of skin diseases linked to connexin gene mutations. Int J Dermatol. 2014; 53: 192‑205. | Crossref
- Belmar P, Marquet A, Martín‑Sáez E. Symmetric palmar hyperkeratosis and esophageal carcinoma [in Spanish]. Actas Dermosifiliogr. 2008; 99: 149‑150. | Crossref
ARTICLE INFORMATION