Coagulation factor XI and coronary artery disease: is there room for factor XI inhibitors?
Key words: atherosclerosis, coronary artery disease, factor XI, factor XI inhibitors, myocardial infarction

 CC BY 4.0
CC BY 4.0
Coagulation factor XI and coronary artery disease: is there room for factor XI inhibitors?
A growing body of evidence indicates that coagulation factors may exhibit not only prothrombotic but also proatherogenic effects. Recent years have yielded numerous animal studies demonstrating that factor (F) XI, largely genetically determined, which constitutes a junction between the tissue factor–dependent coagulation and contact pathways, may contribute to the development and progression of atherosclerosis. Moreover, abundancy of FXI may propagate thrombus formation and render dense and lysis‑resistant fibrin clots, thus contributing to thromboembolic events, such as myocardial infarction (MI) and, in particular, ischemic stroke. Among FXI inhibitors, asundexian was evaluated in patients following acute MI in a phase 2 randomized PACIFIC AMI trial, without clear benefits from this intervention, while the phase 3 LIBREXIA ACS study, which aimed to assess milvexian on top of dual antiplatelet therapy in the prevention of secondary ischemic events in high‑risk MI survivors, was discontinued after interim analysis on the basis of futility. The current review summarizes the latest preclinical and clinical studies on the role of FXI in atherogenesis and its thromboembolic manifestations. Moreover, it discusses potential therapeutic options offered by FXI inhibitors.
Introduction
Coronary artery disease (CAD), encompassing acute and chronic coronary syndromes, remains the leading cause of morbidity, disability, and mortality worldwide.1-3 CAD is largely caused by the formation of atherosclerotic fibrofatty lesions in the coronary arteries and often in other medium and large arteries, which may result in life‑threatening thromboembolic manifestations, such as myocardial infarction (MI) and stroke.4 Atherosclerosis is traditionally associated with dyslipidemia, hypertension, diabetes, obesity, smoking, unbalanced diet, and a sedentary lifestyle.5,6 As stated by Yusuf et al7 based on the INTERHEART study (A Global Study of Risk Factors for Acute Myocardial Infarction), “Abnormal lipids, smoking, hypertension, diabetes, abdominal obesity, psychosocial factors, consumption of fruits, vegetables, and alcohol, and regular physical activity account for most of the risk of myocardial infarction worldwide in both sexes and at all ages in all regions.”7 However, a number of nonconventional contributors to atherosclerosis have been proposed, including particulate matter in air pollution,8 gut dysbiosis,9 or incorrect sleep patterns.10
A chronic, low‑grade inflammatory state, largely driven by the accumulation of oxidized low‑density lipoproteins (LDL) and LDL aggregates within the arterial wall, is acknowledged as the prevailing mechanism behind atherosclerosis.4,11 Additional proatherogenic mechanisms, such as enhanced neutrophil extracellular traps activation and release, oxidative stress, and cellular senescence have also been shown as potent contributors to atherogenesis.11-13
There is controversy around the concept linking atherosclerosis with blood coagulation apart from thromboembolic manifestations of atherosclerotic disease, in particular MI.14,15 Growing evidence indicates that factors involved in blood coagulation and fibrinolysis may modulate the development and progression of atherosclerosis at its early and late stages.16,17 The most relevant contributor appears to be tissue factor (TF), the key initiator of in vivo blood coagulation, abundantly expressed in macrophages, smooth muscle cells, and extracellular vesicles shed from macrophages and cellular components of the atherosclerotic plaque, with a large pool of TF in the acellular necrotic core of the unstable atherosclerotic plaques prone to rupture.18 Apart from triggering factor (F) VII activation and subsequent downstream thrombin formation, the TF‑FVIIa complex acts through protease‑activated receptors (PAR)-1 and 2, enhancing chemotaxis, inflammation, vascular smooth muscle cell migration, proliferation, angiogenesis, and promoting apoptosis.16,18 Previous studies have demonstrated that hallmarks of atherosclerotic progression, such as transformation of macrophages to foam cells, exposure to oxidized LDL, or the formation of the necrotic core are associated with an upregulation of TF expression within atherosclerotic lesions in a manner dependent on nuclear factor κB.19
The contribution of coagulation factors, foremostly TF, FX, thrombin, and fibrin(ogen) to atherosclerosis has been elegantly summarized by Borisoff et al in 2011.16 Since then, a number of studies expanded the knowledge also on the involvement of the contact coagulation pathway, especially FXI, in the progression of atherosclerosis from early lesions to atherothrombotic events. Several reviews have focused on FXI inhibition in the prevention and treatment of venous thromboembolism in patients undergoing surgery and prevention of arterial thromboembolism in atrial fibrillation.20,21 The same holds true for a broad spectrum of cardiovascular diseases due to ongoing FXI‑lowering clinical trials. This review aims to summarize the current basic and clinical evidence on the role of FXI as a modulator of coronary atherosclerosis and atherothrombotic events specifically in CAD. We also provided an update on trials assessing potential FXI inhibition in acute MI and future perspective in this regard.
Factor XI in blood coagulation
FXI is a homodimeric protease with a mass of 160 kDa, synthesized in the liver and circulating in blood at a concentration of 15–45 nM, forming a complex with high‑molecular‑weight kininogen.22 The gene encoding FXI, located on chromosome 4, originates from the duplication of the KLKB1 gene for kallikrein, which makes its structure distinct from other coagulation factors.23 An FXI monomer consists of 4 apple domains (A1–A4) and 1 catalytic (serine protease) domain, with well‑defined molecular partners which can interact with each of these domains.23 FXI may be activated to FXIa by FXIIa, thrombin, or due to autoactivation facilitated by polyanions.22 FXIa activity is physiologically inhibited by several natural inhibitors: C1‑inhibitor, antithrombin, α-1 antitrypsin (α-1AT), protease nexin 1 and 2, and protein Z–dependent protease inhibitor.23,24
The activated form of FXI, FXIa, constitutes a junction between the TF‑dependent coagulation and contact pathways, and is fundamental for sustained thrombin generation and clot amplification.22,25 Due to its contribution to thrombin generation, FXIa indirectly elevates thrombin‑activatable fibrinolysis inhibitor levels, as shown by von dem Borne et al.26 FXIa may cleave a number of substrates from outside the coagulation pathways, such as proteins involved in inflammation (complement factor H, prochemerin, β2 glycoprotein I), regulation of the kallikrein‑kinin system (high‑molecular‑weight kininogen), or platelet activation (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 [ADAMTS13]).23
The expression of FXI is largely determined by genetic variability.27 Associations between CAD risk factors and FXI levels are unclear,28 however, some studies have shown increasing FXI levels with age and body mass index.29-31
FXI deficiency is a rare, mild bleeding disorder with an average prevalence of 1 per million in the general population,32 and may be caused by one of the currently known at least 190 mutations within the F11 gene.33 Regarding acquired deficiency, FXI may also be decreased in cirrhosis34 or sometimes in nephrotic syndrome.35
Determination of factor XI
The most common, routine 1‑stage clotting assay is used to determine FXI in everyday practice, however, a few methods have been introduced for research purposes to determine FXI levels and / or activity, in particular immunoenzymatic methods to assess FXI antigen or FXIa in complexes with its inhibitors, and monoclonal antibody–based coagulometric assays.
The 1‑stage activated partial thromboplastin time (aPTT)-based factor XI activity assay is the most commonly available, standardized, and automated assay developed to diagnose FXI deficiency by assessing the ability of a tested sample to correct the aPTT of a standard, FXI‑deficient plasma.36 The aPTT of the tested sample is measured and plotted against standard curves. The differences in aPTT of the dilutions of standard plasma and the tested plasma allow for calculating the patient’s FXI activity level, as a measure of FXI concentration.36 The reference range of FXI coagulant activity (FXI:C) varies in the literature, from 65–125 U/dl to 70–150 U/dl.37
The chromogenic 2‑stage factor XI activity assay comprises 2 steps: the first step, where kaolin‑activated FXII converts FXI into FXIa, and the second step, where p-nitroaniline is added and cleaved by FXIa at the C‑terminus, generating a chromogenic product, which is measured photometrically. The change in optical density of the sample is proportional to FXI concentration. This assay is automated but approved solely for research purposes.37
A number of enzyme‑linked immunosorbent assays (ELISAs) are available for the measurement of FXI antigen level in the plasma or serum for research purposes, among which the kits from Affinity Biologicals (Ancaster, Canada) and 5‑Diagnostics (Basel, Switzerland) are traceable to the World Health Organization international standard for FXI antigen in human plasma and the International Society on Thrombosis and Haemostasis, Scientific and Standardization Committee standard for FXI activity, respectively.37
In 1995, Wuillemin et al38 published a reference study showing that in human plasma almost half (47%) of FXIa forms complexes with C1 inhibitor, followed by α2‑antiplasmin (24.5%), α1AT (23.5%), and antithrombin III (5%). Importantly, these proportions remained unchanged regardless of whether FXIa was added to plasma, or activated endogenously by negatively‑charged molecules, with marginal variability at lower temperatures or in the presence of heparin.38 Based on the assumption that FXIa is neutralized by its natural inhibitors in constant proportions, the amount of FXIa may be estimated by ELISA measurement of its complexes with C1 inhibitor or α1AT.39-41
Further, Minnema et al42 described an assay designed for the selective detection of FXIa, where the enzyme is captured on a plate covered with a specific antibody, followed by an addition of a chromogenic substrate of FXIa. Absorbance values allow for a calculation of molar concentrations of FXIa, provided the molecular weight of a FXIa monomer (80 kDa).42 Interestingly, peak values of noncomplexed FXIa measured using this method preceded peak FXIa‑inhibitor complex levels by several hours, suggesting that FXIa‑inhibitor binding takes place on cellular surfaces and not in the plasma compartment.42
Another method for a selective FXIa measurement, introduced by Butenas et al,43 is based on the properties of αFXI inhibitory monoclonal antibodies targeting FXI/FXIa, which are added to citrated plasma, preventing FIX activation by FXIa. This is followed by an addition of calcium ions and phospholipid vesicles to initiate coagulation. Clotting times are measured and calculated based on calibration curves obtained from measurements of a standard sample of human FXIa. The detection level for FXIa in plasma using this technique is 10 pM. FXIa has not been detected in healthy individuals using this approach.43
Recently, a novel, multivalent catch‑and‑release assay, based on a naturally occurring FXIa inhibitor fasxiator and biotin, has been proposed to measure circulating FXIa with a detection limit of 20 pM in plasma.44
Factor XI and coronary artery disease: preclinical and animal studies
Coagulation factors, including FXI, are abundant in early‑stage atherosclerotic lesions, with a 3‑fold higher activity of TF and FXa, and 2‑times increased FXII activity in early vs stable advanced lesions, especially in the vicinity of macrophages and smooth muscle cells, suggesting a procoagulant state that may serve as a “defense” mechanism to stabilize the newly‑forming plaque.15,45 As coagulation factors enhance inflammation, proliferation, and angiogenesis, their local upregulation within the vessel wall may gradually lead to a vicious circle of events that drive the progression of atherosclerosis, as shown in a number of animal studies.45
It has been reported that mice with a double apolipoprotein E (ApoE) and FXI knockout had significantly reduced atherosclerotic lesion area measured in the aortic sinus and arch, as compared with ApoE knockout mice with normal FXI expression, despite no differences in lipid levels between the animals.46 Another report using the same model showed that in comparison with ApoE–/– mice, animals with an additional FXI knockout had a lower plaque monocyte infiltration and exhibited downregulation of the macrophage scavenger receptor 1 (MSR1) gene, the product of which is a strong proinflammatory and proatherogenic factor.47,48 In LDL‑receptor deficient mice on a high‑fat diet, the administration of 14E11, an antibody targeting FXI, or a FXI antisense oligonucleotide (FXI‑ASO) resulted in a reduction of lesion area in the aorta.49 The same study provided an observation that cultured human umbilical vein endothelial cells exposed to FXIa, but not inactive FXI, exhibited disruptions in vascular endothelial (VE)-cadherin expression and increased endothelial permeability to acetylated LDL.49 Puy et al50 also reported FXIa‑driven VE‑cadherin cleavage in human venous and aortic endothelial cells, in a process regulated by disintegrin and metalloproteinase 10, which resulted in increased permeability of the monolayer.
FXI was shown to exhibit proinflammatory properties, likely due to its ability to activate components of the kallikrein‑kinin system. FXIa may release bradykinin from high‑molecular‑weight kininogen.51 FXI‑deficient mice with induced sepsis showed lower levels of proinflammatory cytokines, in association with lower mortality.52 Administration of a humanized anti‑FXI monoclonal antibody, h1A6, in rhesus macaques with hyperlipidemia reversed several hallmarks of thromboinflammation: decreased C‑reactive protein levels, attenuated platelet glycoprotein VI and PAR‑1–dependent platelet activation, and lowered vascular cell adhesion molecule‑1 expression in the endothelium.53 In line, a recent proteomic study performed in patients with venous thromboembolism showed that both in the acute phase and long‑term observation, elevated FXI was associated with upregulation of proteins involved in thromboinflammation and lipid oxidation, along with apoptosis, extracellular matrix degradation, and protein misfolding.54
A growing number of experimental studies demonstrate that FXI plays a role in sustaining thrombin generation, once clotting has been initiated. In mice with chemically induced atherothrombosis within the carotid artery, FXI deficiency was associated with decreased thrombus formation.55 Consistently, an infusion of wild‑type mice with the 14E11 antibody targeting FXI activation showed a similar inhibition of thrombus formation.56 Also, van Montfoort et al57 showed that pretreatment of ApoE–/– mice with FXI‑ASO resulted in slower formation of smaller and unstable thrombi, along with lower fibrin deposition 5–10 minutes after the induction of carotid atherothrombosis. Importantly, plaque collagen content and macrophage infiltration were lower in the FXI‑ASO treated animals.57 In a primate model where a collagen‑coated graft was introduced into an arteriovenous shunt to stimulate thrombosis, administration of 14E11 reduced fibrin and platelet accumulation within the graft and prevented thrombus growth.58 Infusion of FXI‑ASO in the same model led to a decrease in clot growth along with inhibition of thrombin generation, suggesting that thrombus propagation was largely dependent on FXI‑derived thrombin in this model.59 Finally, preliminary studies suggest that FXI may enhance platelet aggregation by inactivating ADAMTS13, the metalloproteinase attenuating von Willebrand factor,60 and by supporting thrombin generation on platelet surface via receptor glycoprotein Ibα.61 Higher availability of FXI/FXIa in human plasma was shown to enhance thrombin generation and negatively affect fibrin clot properties, increasing clot density and impairing its lysability.62,63 Such a “prothrombotic fibrin clot phenotype” in CAD was shown to be an independent predictor of atherothrombotic events, including MI.63,64
Although head‑to‑head comparisons between direct oral anticoagulants and FXI inhibitors are unavailable in CAD, given the anticoagulant potency of FXa and thrombin inhibition, well‑documented in both preclinical studies and clinical trials,65,66 it seems that the relative potency of FXI inhibition in preventing thromboembolism in CAD may be modest, especially when compared with direct thrombin blockage. Nevertheless, FXI attenuation may still have potential in settings, where direct thrombin or FXa inhibition is associated with an unacceptably high bleeding risk.
Taken together, evidence shows that FXI may exacerbate atherosclerosis by augmenting inflammation, and increasing endothelial permeability to leukocytes and LDL. In models of atherothrombosis, FXIa is a contributor to thrombus propagation and stability (Figure 1). Results of preclinical and animal studies are detailed in Table 1.
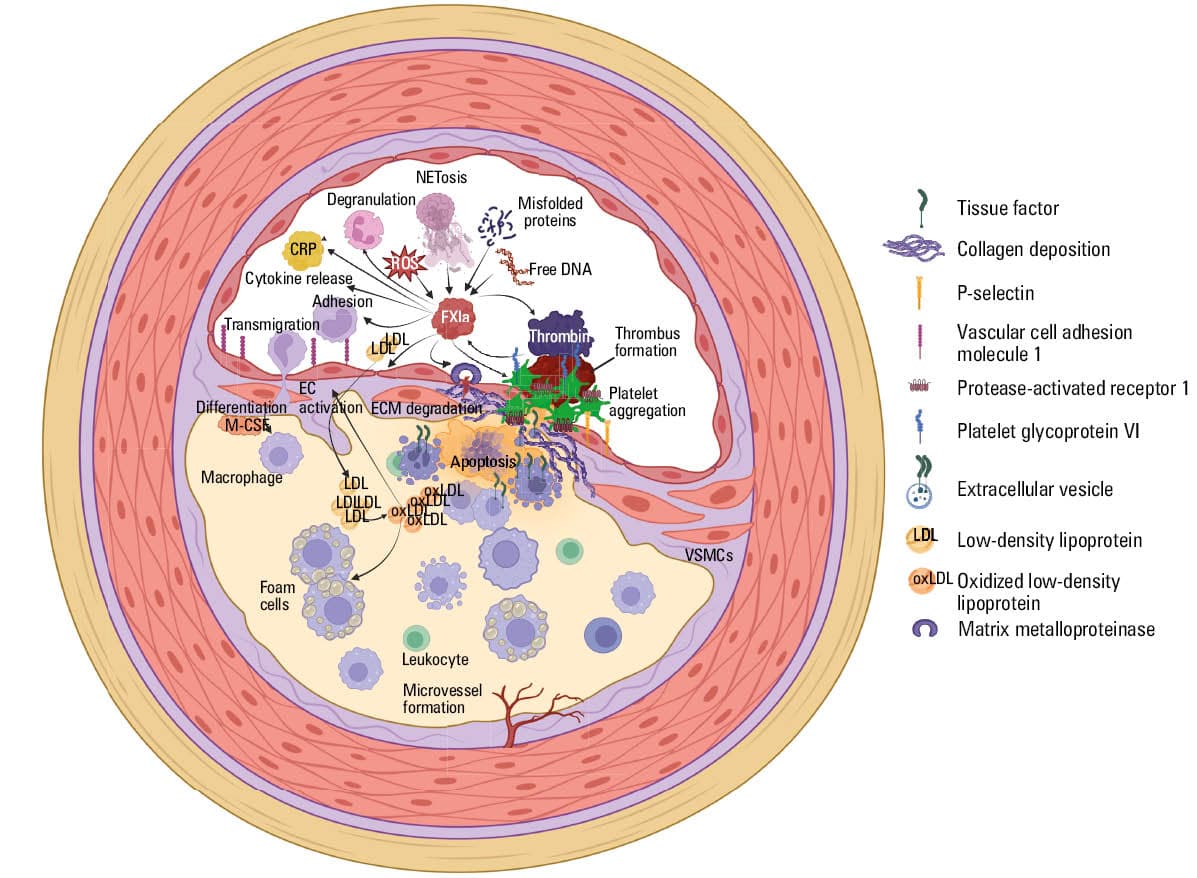
Abbreviations: CRP, C‑reactive protein; ECM, extracellular matrix; FXIa, activated factor XI; M‑CSF, macrophage colony‑stimulating factor; NETosis, neutrophil extracellular trap activation and release; ROS, reactive oxygen species; VSMC, vascular smooth muscle cell
Author, year, reference | Model | Outcome |
Abbreviations: ↓, decrease; ↑, increase; ADAMTS13, disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; ApoE–/–, apolipoprotein E knockout; ASO, antisense oligonucleotide; DKO, double knockout; FXI, factor XI; GPVI, platelet glycoprotein VI; KO, knockout; LDL, low‑density lipoprotein; MRS1, macrophage scavenger receptor 1; PAI‑1, plasminogen activator inhibitor‑1; PAR‑1, protease‑activated receptor‑1; VCAM‑1, vascular cell adhesion molecule‑1; VE, vascular edothelial; VEGFR2, vascular endothelial growth factor receptor 2; VLDL, very low‑density lipoprotein; VLDLR, very low‑density lipoprotein receptor; WT, wild‑type; others, see Figure 1 | ||
Shnerb Ganor, 201646 | ApoE/FXI DKO mice vs ApoE –/– only mice | ApoE –/–, FXI –/– DKO mice vs ApoE KO mice: ↓ 25% (P = 0.02) atherosclerotic lesion area in the aortic sinus and ↓ 49% (P = 0.03) in the aortic arch; ↓ macrophage infiltration |
Ngo, 202149 | Ldlr –/– mice on high‑fat diet + 14E11 or FXI‑ASO | ↓ Lesion area in the proximal aorta by 39% (14E11) and 33% (FXI‑ASO) |
Ngo, 202149 | Human endothelium cultured with FXIa | ↑ Permeability to LDL |
Ngo, 202149 | Ldlr–/– mice exposed to FXI and FXI inhibitor | Disruption of endothelial barrier in aortic lesions; no endothelial dysfunction with 14E11 anti FXI |
Shnerb Ganor, 202547 | ApoE –/– vs ApoE –/– and FXI–/– DKO mice on high‑fat diet, RNA expression in atheroma at 24 weeks | ↓ MRS1 RNA (98 vs 3575 RNA copies; P = 0.002) in ApoE–/– FXI–/– vs ApoE–/– only mice |
Puy, 202450 | Human umbilical endothelial cells in vitro incubated with FXIa | ↓ VE‑cadherin, ↑ endothelial permeability, ↑ ADAM10 expression and activity;
FXIa–PAI‑1 – endothelial VLDL receptor interactions: ↑ cell signaling, and ↓ VE‑cadherin |
Puy, 202450 | Human aortic endothelial cells in vitro incubated with FXIa | VE‑cadherin ↓ through ADAM10, VEGFR2, and VLDLR |
Kohs, 202453 | Rhesus macaques with hyperlipidemia on high‑fat diet treated with h1A6, an anti‑FXI antibody | ↓ P‑selectin expression in response to GPVI and PAR‑1 activation, ↓ VCAM‑1 expression in the endothelium, ↓ CRP |
Cheng, 201056 | FXI‑deficient mice vs WT mice infused with 14E11, an antibody targeting FXI, FeCl3-induced carotid thrombosis | ↑ Time to occlusion |
Van Montfoort, 201457 | ApoE –/– mice with ultrasound‑induced carotid artery thrombosis, pretreated with FXI‑ASO | 5–10 min post clotting initiation: ↓ thrombus formation, ↓ thrombus size (mean [SD],14.1 [4] × 103 μm2 in FXI‑ASO group vs 68.4 [23.9] × 103 μm2 in placebo group; P <0.05), ↓ fibrin deposition, ↓ platelet aggregate stability, within plaques: ↓ macrophage infiltration, ↓ collagen deposition |
Cheng, 201056 | Baboon, arteriovenous shunt with a collagen‑coated graft introduced to evoke thrombosis, pretreatment with 14E11, an antibody targeting FXI | ↓ Thrombus growth within graft, no effect on platelet deposition |
Crosby, 201359 | Baboon, arteriovenous shunt with a collagen‑coated graft introduced to evoke thrombosis, pretreatment with ASO targeting FXI | Near‑complete inhibition of thrombus formation within graft at >80% blocked FXI; ↓ thrombin generation; no effect on platelet aggregation |
Kossmann, 201761 | Normocholesterolemic C57BL/6 mice, infused with angiotensin II, resulting in hypertension and leukocyte transmigration to the artery wall | FXI‑ASO infusion led to ↓ endogenous thrombin potential by 20%, leukocyte infiltration ↓ 58%, ↓ expression of genes involved in endothelial dysfunction (Ccl2, Vcam‑1, Ccr2, Spn) |
Puy, 201760
(conference abstract) | ADAMTS13 incubated with FXIa | ↓ ADAMTS13, loss of ADAMTS13 CUB domain, essential for inactivation of von Willebrand factor |
Factor XI and coronary artery disease: clinical studies
Factor XI deficiency and coronary artery disease
It was suggested that an inborn FXI deficiency may protect against MI and ischemic stroke.67,68 A seminal study by Preis et al68 conducted on more than 10 000 individuals from the general Israeli population showed that a mild or moderate FXI deficiency decreased the probability of cardiovascular events defined as MI, ischemic stroke, or transient ischemic attack. A lower risk of ischemic stroke, but not MI, was shown in an observational study of 115 individuals with a severe FXI deficiency (ie, ≤15 U/dl) and 1528 controls with normal FXI activity.67 Similarly, a severe FXI deficiency did not reduce the risk of MI in a Jewish cohort study of 96 FXI‑deficient patients and the general population.69
Elevated factor XI and stable coronary artery disease
In 1995, a cross‑sectional study by Murakami et al70 showed elevated FXI-α1AT complexes in CAD patients with significant coronary stenoses as compared with those with normal coronary angiography (by 15.8%), as well as healthy controls (by 16.8%), suggesting a link between FXI activity and the severity of coronary atherosclerosis. Persistently elevated FXI, assessed using the 1‑stage clotting technique, along with prekallikrein, and high‑molecular‑weight kininogen, were found in MI survivors in the stable phase of CAD, as compared with healthy controls.71 Interestingly, 59% of the patients had a family history of CAD, which is in line with the fact that FXI levels are determined primarily by genetic variability.71 Women with stable CAD and multivessel disease (defined as ≥50% stenosis in ≥3 coronary arteries) had 56% higher FXI level measured with the 1‑stage technique than those in whom no significant coronary lesions were found (128% vs 82%; P <0.04), and this association was independent of age and classic atherosclerosis risk factors.72
Our analysis, with the method in which monoclonal antibodies against FXIa were employed, showed that FXIa, along with active TF, were detectable in 40% of patients with ischemic cardiomyopathy, in association with younger age and higher prevalence of history of MI,73 indicating that these factors may affect the advancement of coronary atherosclerosis. A link between FXIa and the severity of coronary atherosclerotic lesions has also been shown in patients with stable CAD, likely in association with markers of inflammation and oxidative stress.74
Elevated factor XI and risk of atherothrombotic events
Data on FXI and atherothrombosis, especially MI, vary depending on the population and the method used to assess FXI. In our cohort with stable, advanced CAD followed long‑term, circulating FXIa was common and associated with an over 6‑fold higher rate of a thromboembolic composite end point defined as MI, stroke, systemic thromboembolism, or cardiovascular death.75 Similarly, in patients with well‑controlled type 2 diabetes (64.7% of whom had coexisting CAD and 18.8% were MI survivors), baseline FXI:C above 120% was a predictor of MI, stroke, or cardiovascular death after a median 5‑year observation, and this association remained valid after adjustment for age, sex, and LDL cholesterol.76 In another study, FXI:C was associated with a 1.8‑fold higher risk of recurrent MI in a population of 560 men below the age of 70 years who experienced acute coronary syndrome and 646 controls,77 while there was no such association for 200 female MI survivors aged below 49 years and 626 matched controls.78
On the other hand, a prospective cohort study of individuals without a diagnosis of atherosclerotic disease showed an association between high levels of FXI antigen defined as at least 75th percentile (≥5.92 mg/l) and the occurrence of stroke (odds ratio [OR], 1.66; 95% CI, 1.05–2.65), but not acute coronary syndrome in more than 1800 individuals followed for a median time of 2.7 years.79 The RATIO study (Risk of Arterial Thrombosis in Relation to Oral Contraceptives) including women aged 18 to 50 years who had suffered from MI (n = 205) or stroke (n = 175) and matched healthy controls (n = 638) showed that enhanced FXI activity, measured as complexes of FXIa with its inhibitors, was associated with the risk of stroke (OR for FXIa‑C1‑inhibitor, 2.8; 95% CI, 1.6–4.7 and OR for FXIa‑AT‑inhibitor, 2.3; 95% CI, 1.4–4), but not MI.80 Finally, single‑nucleotide polymorphisms revealed that genetic variability of the F11 gene was associated with stroke risk, attributable to cardioembolic stroke and stroke of undetermined cause, but not large artery atherosclerosis or small artery occlusion.81
Summing up, there is a convincing body of data showing associations between elevated FXI and the risk of thromboembolic events, especially in advanced CAD. The evidence is best delineated for ischemic stroke, while the role in MI occurrence is less clear and seemingly dependent on the type of test used to detect FXI levels or activity, as well as detailed characteristics of the population.
Elevated factor XI during acute myocardial infarction
The first report on detectable FXIa in acute MI (AMI) was published more than 15 years ago, followed by several studies, all of which showed FXI/FXIa elevation with varying levels, depending on the assay and population characteristics. In 2008, Butenas et al43 reported FXIa above 10 pM (mean [SD], 50 [33] pM; range, 16–120 pM) in 96% of a small cohort of Polish patients with AMI admitted to a hospital within 12 hours of symptom onset. In line, Konings et al40 showed elevated FXIa measured as complexes with C1 inhibitor or antithrombin in a Dutch population of 89 patients with AMI. The same study reported that the levels of FXIa with its inhibitors had no predictive value in MI survivors during a 1‑year observation.40 In another small Dutch cohort study (n = 56), mean (SD) FXIa level assessed using an antibody‑based thrombin generation assay was elevated in AMI (3.7 [2.7–5.5] pM), and decreased to levels similar to those measured in healthy controls (2.7 [1.6–4.2] pM) 6 months post the index event.82 Consistently with Konings et al,40 the authors found no predictive value for FXIa level during a 1‑year observation. A more recent German cohort prospective study with 3‑year follow‑up of more than 570 stroke survivors showed an association between FXI:C above 75th percentile measured using the 1‑stage clotting technique and a risk of recurrent stroke, MI, or death (hazard ratio [HR], 1.8; 95% CI, 1.09–2.98).83
Collectively, small observational studies show that the acute phase of MI is accompanied by a rise in FXI antigen and activity, however, its levels during this phase do not seem to predict the risk of recurrent events in long‑term observation.
Factor XI inhibitors in coronary artery disease
Addition of FXI inhibitors to standard therapy in CAD, not in monotherapy, could be a therapeutic option, especially for high‑risk CAD patients with elevated FXI levels. Given the marginal role of FXI in hemostasis, treatment with an FXI inhibitor on top of antiplatelet therapy could reduce disease progression20 with a negligible increase in clinically relevant bleeding, which is of key importance, especially in the elderly predominantly at moderate‑to‑high bleeding risk.84
To date, several classes of FXI inhibitors have been proposed, including antibodies, antisense oligonucleotides, small molecules, aptamers, and natural inhibitors (Figure 2).23,85 However, few randomized controlled trials (RCTs) have been designed to test FXI inhibitors in CAD in contrast to venous thromboembolism prevention or atrial fibrillation.
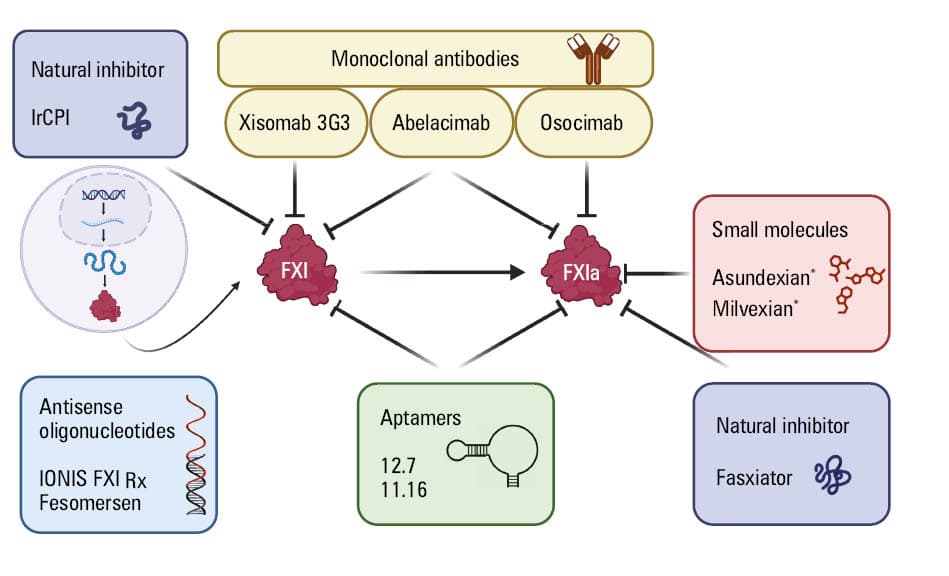
Abbreviations: IrCPI, Ixodes ricinus contact phase inhibitor; others, see Table 1 and Figure 1
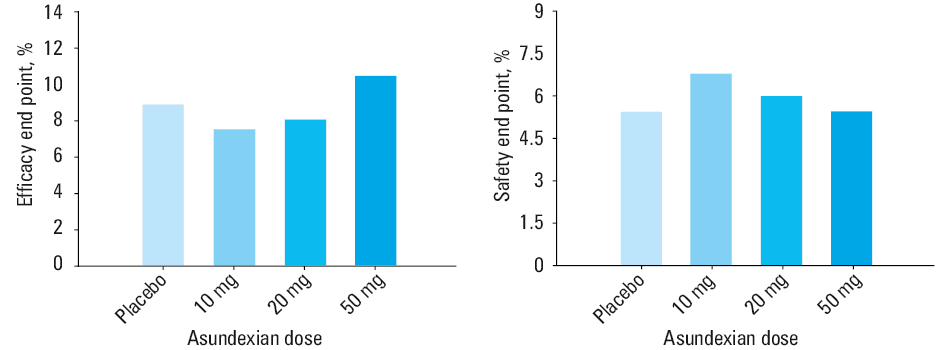
So far, the only phase 2 RCT performed in CAD was the PACIFIC AMI (Study to Gather Information about the Proper Dosing and Safety of the Oral FXIa Inhibitor BAY 2433334 in Patients Following an Acute Heart Attack; NCT04304534), aiming to assess the pharmacodynamics, safety, and efficacy of asundexian in individuals with recent MI.86 The trial was conducted on 1601 patients, who were randomized up to 5 days post the acute event to receive 10, 20, or 50 mg oral asundexian once daily, or placebo, in addition to dual antiplatelet therapy for a period of 6 or 12 months. The mean patient age was 68 years, and 1 in 5 participants was a woman. Acute coronary syndrome manifestation was ST‑segment elevation myocardial infarction in 51% of cases. Nearly all patients (99%) were treated with percutaneous angioplasty and 80% received aspirin and ticagrelor / prasugrel thereafter. As assessed after 4 weeks, asundexian caused a dose‑dependent attenuation of FXI activity, with a more than 90% inhibition for 50 mg, as compared with placebo.86 Over a median 1‑year follow‑up period, no differences were found in the safety outcome defined as grade 2, 3, or 5 bleeding in the Bleeding Academic Research Consortium with a HR of 0.98 and 90% CI, 0.71–1.35 for pooled asundexian vs placebo. There was no change in the efficacy end point defined as MI, in‑stent thrombosis, stroke, or cardiovascular death (HR, 1.05; 90% CI, 0.69–1.6 for pooled 20 and 50 mg asundexian vs placebo), but the study was underpowered to detect differences in this regard (Figure 3).86 Subsequently, the phase 3 trial to test asundexian in AMI patients has not been planned.
The phase 3 LIBREXIA ACS (A Study of Milvexian in Participants After a Recent Acute Coronary Syndrome; NCT05754957) RCT,87 which aimed to assess the efficacy and safety of milvexian in the prevention of cardiovascular death, recurrent MI, or ischemic stroke in MI patients with at least 2 features of a high risk of ischemic events, has recently been stopped following interim analysis “due to insufficient conditional power to achieve the study’s primary objective.” Of note, high ischemic risk criteria used for enrollment in this study included unmodifiable factors, such as complicated coronary anatomy or prior coronary interventions. The risk associated with such features is unlikely to be attenuated with FXI/FXIa inhibition. However, it is tempting to suggest that the measurement of FXI levels in CAD patients, especially those with a simultaneously high ischemic and bleeding risk could help identify suitable recipients of FXI‑targeted therapies.
Taken together, the present findings on FXI inhibition indicate that such an intervention has a small effect on clinically relevant ischemic events in CAD, which might be at least partly related to dosage regimens, patient characteristics, and cointerventions. As in the case of combination therapy of low‑dose rivaroxaban and aspirin,88 FXI inhibitors at appropriate daily doses could be more effective in patients at a high risk of cardiovascular events, but not in acute ischemia. Further research is needed to optimize the use of FXI inhibitors in CAD patients.
Future perspectives
There is evidence indicating that FXIa may enhance atherosclerosis at least partly by augmenting inflammation and promoting fibrin formation. Given the rapidly evolving experimental and clinical studies, the next decade will likely elucidate the mechanisms through which FXI may be implicated in the development of early atherosclerotic lesions, their progression, and finally atherothrombotic events. Based on the role of FXI/FXIa in thrombin amplification and thrombus propagation, currently it seems that FXI inhibitors could have a place in the secondary prevention of thromboembolic events in selected patients with a high ischemic risk attributable to elevated FXI levels, especially with a concomitant high bleeding risk. It is tempting to suggest that if the regulatory role of FXIa in early atherogenesis is confirmed, FXI inhibitors could be used in the prevention of CAD progression.
- Roth GA, Mensah GA, Johnson CO, et al. Global burden of cardiovascular diseases and risk factors, 1990‑2019: update from the GBD 2019 study. J Am Coll Cardiol. 2020; 76: 2982‑3021. | Crossref
- Kardas P, Kwiatek A, Włodarczyk P, et al. Is the KOS‑Zawał coordinated care program effective in reducing long‑term cardiovascular risk in coronary artery disease patients in Poland? Insights from analysis of statin persistence in a nationwide cohort. Pol Heart J. 2024; 82: 852‑860. | Crossref
- Szum‑Jakubowska A, Chlabicz M, Dubatówka M, et al. Cardiovascular risk and preclinical atherosclerosis are associated with white matter hyperintensities in apparently healthy adults: the population‑based cross‑sectional BIALYSTOK PLUS study. Pol Arch Intern Med. 2024; 134: 16825. | Crossref
- Libby P. The changing landscape of atherosclerosis. Nature. 2021; 592: 524‑533. | Crossref
- Brown JC, Gerhardt TE, Kwon E. Risk Factors for Coronary Artery Disease. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. https://www.ncbi.nlm.nih.gov/books/NBK554410/. Accessed November 7, 2025.
ARTICLE INFORMATION