Chronic lymphocytic leukemia mimicking lung carcinoma: an unusual extranodal and extramedullary presentation of a frequent hematological neoplasm

 CC BY 4.0
CC BY 4.0
Chronic lymphocytic leukemia mimicking lung carcinoma: an unusual extranodal and extramedullary presentation of a frequent hematological neoplasm
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia in Western countries.1 Although its clinical course is often indolent at onset, CLL may present with extranodal and extramedullary manifestations, most frequently in the liver. The nonlymphatic system involvement is rare, with the most prevalent sites being the skin and the central nervous system.2 Pulmonary involvement is uncommon and may occur due to leukemic pulmonary infiltration (LPI).
We present a case of a 75‑year‑old man with a history of hyperlymphocytosis and lymphadenopathy since 2017. Bone marrow trephine biopsy demonstrated 70% infiltration with CLL cells. Cytogenetics showed a deletion of the short arm of chromosome 17 (del[17p]) mutation in 3.7% of the cells. In 2020, the patient received first‑line chemoimmunotherapy (fludarabine, cyclophosphamide, and rituximab regimen), achieving partial remission.3 The patient remained in observation for approximately 18 months, after which he presented with facial edema, fatigue, night sweats, and pleuritic chest pain. Chest computed tomography (CT) identified a centrally located mass in the right lung (Figure 1A and 1B). Positron emission tomography / CT confirmed a metabolically active lung mass, which was interpreted as likely CLL infiltration with a possible inflammatory component (Figure 1C–1F).
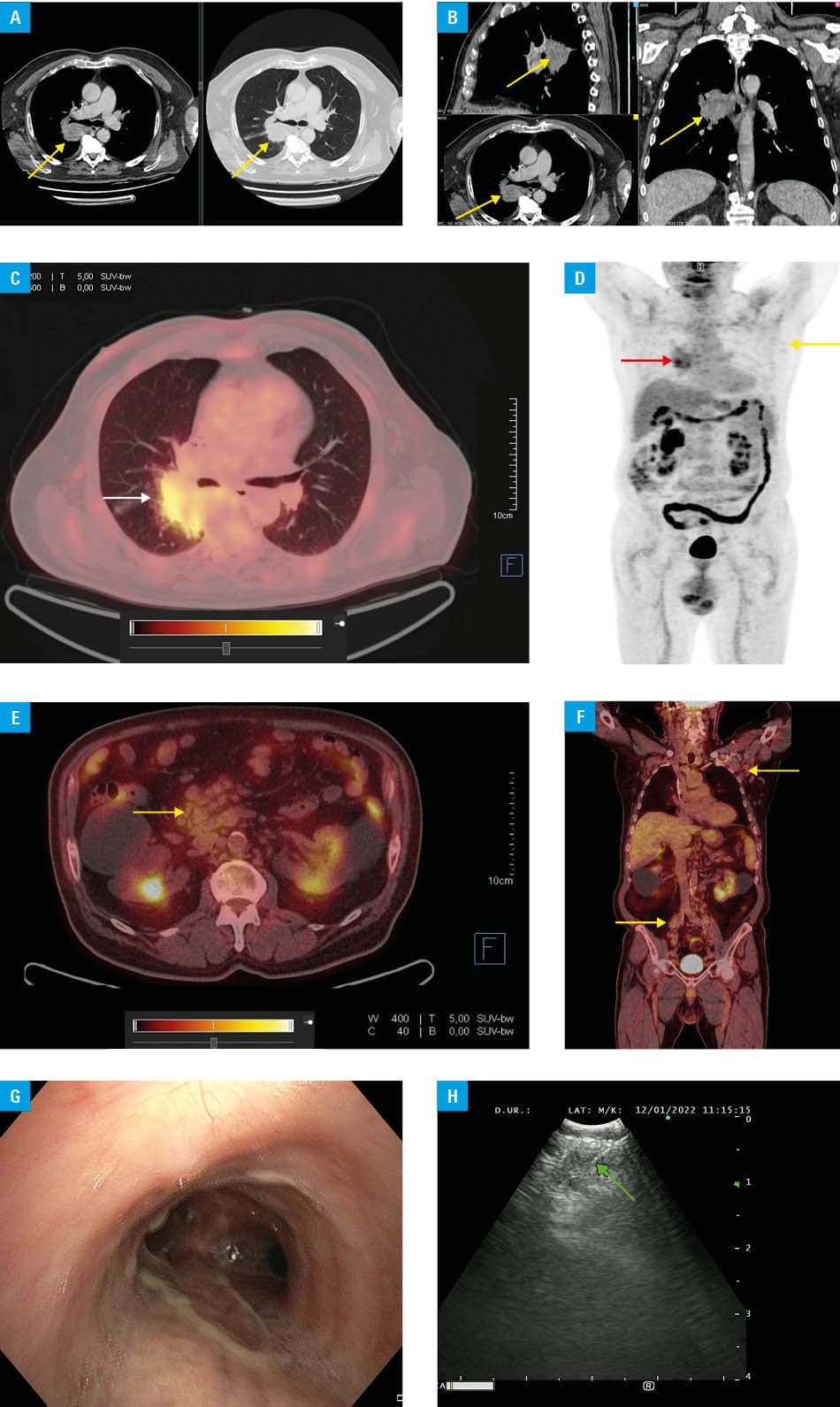
Bronchofiberoscopy with endobronchial ultrasound–transbronchial needle aspiration (EBUS‑TBNA) of the pulmonary lesion and mediastinal nodes (Figure 1G and 1H) yielded lymph node tissue fragments with small lymphoid cells (Pa × 5+/−, CD5+, CD23−/+, cyclin D1−, and partial CD3+), accompanied by histiocytes. These findings were consistent with LPI, although immunophenotyping was limited by low cellularity.
A genetic analysis of the peripheral CLL cells excluded a del(17p) or tumor protein p53 (TP53) mutation at this stage. Treatment with venetoclax and rituximab was initiated.4 Six months later, CT demonstrated significant regression of the lung mass and complete resolution of lymphadenopathy. After the completion of therapy, only a fibrotic scar was found on CT. At the time of this report, the patient remained in remission,3 although bone marrow re‑evaluation to confirm complete remission had not been performed.
This case highlights LPI as a rare but important differential diagnosis in CLL patients presenting with mass‑like pulmonary lesions. Several mechanisms causing LPI have been proposed—including direct leukemic infiltration along lymphatic routes, peribronchovascular spread, and leukostasis—in the cases of extreme lymphocytosis. Histopathological patterns described in the literature range from diffuse interstitial infiltration to perilymphatic nodules. Mass‑forming lesions resembling lung cancer are exceedingly rare. Moreover, most published cases rely on surgical biopsy, whereas in our patient, the diagnosis was established with minimally‑invasive EBUS‑TBNA. The combination of a centrally located tumor‑like lesion, coexistence of TP53 abnormalities at initial diagnosis, and a marked response to venetoclax and rituximab further distinguishes this case from previously published reports.
Radiologic features of LPI may be indistinguishable from primary lung cancer, particularly given the increased risk of second primary malignancies in CLL patients. In addition, Richter transformation (RT), infections, or therapy‑related changes need to be considered. In RT, lesions typically demonstrate high metabolic activity, rapid growth, and systemic deterioration—none of which were present in our patient. Therefore, histologic verification is essential, with large B‑cell lymphoma (BCL) morphology being typical of RT. Recognition of LPI is crucial to avoid unnecessary surgical interventions and ensure timely initiation of appropriate therapy.
The favorable response achieved with venetoclax and rituximab underscores the importance of novel therapies, including targeted BCL‑2 inhibition, in treating CLL patients with extranodal or extramedullary involvement, such as LPI.
- Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. 2022; 36: 1720‑1748. | Crossref
- Robak T, Pula A, Braun M, Robak E. Extramedullary and extranodal manifestations in chronic lymphocytic leukemia ‑ an update. Ann Hematol. 2024; 103: 3369‑3383. | Crossref
- Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018; 131: 2745‑2760. | Crossref
- Kater AP, Harrup R, Kipps TJ, et al. The MURANO study: final analysis and retreatment / crossover substudy results of VenR for patients with relapsed / refractory CLL. Blood. 2025; 145: 2733‑2745. | Crossref
ARTICLE INFORMATION