Elevated factor XI activity is associated with enhanced neutrophil extracellular trap formation in patients with deep vein thrombosis: a post hoc analysis of a cohort study

 CC BY 4.0
CC BY 4.0
Elevated factor XI activity is associated with enhanced neutrophil extracellular trap formation in patients with deep vein thrombosis: a post hoc analysis of a cohort study
Introduction
There is compelling evidence for the role of neutrophils in thrombus formation, particularly through the release of neutrophil extracellular traps (NETs) by activated neutrophils, a process known as NET formation (NETosis).1 During NETosis, activated neutrophils release DNA and granular proteins that stabilize the clot by promoting platelet aggregation and fibrin deposition, while simultaneously impairing clot resolution. NETosis can be assessed by determining the level of, among others, citrullinated histone H3 (H3cit), which is considered a relatively specific marker.2,3 NETosis may contribute to the development and persistence of venous thromboembolism (VTE).4 Recently, we have demonstrated in 179 patients with deep vein thrombosis (DVT) that each 1 ng/ml increase in H3cit levels at 3 months was independently associated with the subsequent development of post‑thrombotic syndrome (PTS; odds ratio [OR], 5.5; 95% CI, 2.52–12.01).5 Moreover, higher H3cit levels correlated with denser fibrin clots and impaired fibrinolysis, which implicate NET‑driven fibrin clot abnormalities as a novel mechanism underlying PTS, a common complication of VTE that affects up to 50% of patients.5
Coagulation factor XI (FXI) links contact activation to thrombin generation,6 and elevated FXI activity is a well‑established independent risk factor for DVT.7,8 Regarding PTS, FXI levels equal to or above 120% were associated with a 5.55‑fold increased risk of PTS independently of other risk factors (OR, 5.55; 95% CI, 2.28–13.47).9
In this study, we tested a hypothesis that elevated H3cit levels are associated with increased FXI activity in DVT patients, and that these 2 mechanisms may act synergistically to heighten the risk of developing PTS. Such an association may help stratify individuals according to their likelihood of experiencing PTS within 1–2 years after the thrombotic event.
Patients and methods
We studied 172 white patients aged 18–69 years with a first episode of isolated DVT or DVT with pulmonary embolism confirmed on imaging.10 This group was a subset of a previously studied population.5,9,11 In brief, the patients were eligible, if they were on anticoagulation for at least 3 months without DVT progression or recurrence. Key exclusion criteria were severe thrombophilia, acute arterial thromboembolism within the last 3 months, known cancer, C‑reactive protein (CRP) level above 15 mg/l, diabetes mellitus, and advanced chronic kidney disease stage 4 or 5.
The patients were treated with anticoagulants for at least 12 weeks in provoked cases, and for at least 6 months in unprovoked cases, as described. Definitions of all conditions were presented previously.11
The Kraków Medical Chamber Ethics Committee approved the study (135/KBL/OIL/2013), which was conducted in compliance with the Declaration of Helsinki. Written informed consent was obtained from all participants.
The presence of PTS was assessed using the Villalta scale at 1 year after DVT, with a score equal to or above 5 on 2 visits (≥3 months apart) confirming diagnosis. Symptomatic VTE recurrence was recorded and confirmed on compression ultrasound or computed tomography angiography, as described. Follow‑up continued for up to 76 months to diagnose venous ulcers, with visits every 6 months.11
Laboratory investigations
The patients on vitamin K antagonists (n = 170; 98.8%) were switched to a low‑molecular‑weight heparin (LMWH) for 10–14 days, with samples taken when the international normalized ratio was below 1.2, and at least 12 hours after the last LMWH injection. Lipid profile, glucose, creatinine, fibrinogen, and CRP were measured using standard methods.
H3cit level was quantified with an enzyme‑linked immunosorbent assay kit (Cayman Chemical, Ann Arbor, Michigan, United States), with a reference range of 0.5–1.7 ng/ml.5,9,11 Plasma FXI activity was determined with a 1‑stage clotting assay using FXI‑deficient plasma (Siemens, Marburg, Germany) with a reference range of 70%–120%. Elevated FXI activity was defined as values equal to or above 120%.9
The participants were stratified into 2 subgroups according to the quartile distribution of FXI activity and H3cit levels. The first subgroup included patients in the highest quartile for both markers, whereas the second comprised all other participants.
Statistical analysis
Normality was assessed using the Shapiro–Wilk test. Variables with a normal distribution are presented as mean and SD, while those without a normal distribution are presented as median and interquartile range (IQR). Nominal variables are presented as percentages. Differences between the groups were assessed using the t test if the distribution was normal; otherwise, the Mann–Whitney test was applied. For categorical variables, either the Pearson χ2 test or the Fisher exact test was used. Spearman rank correlation coefficient was used to assess the association between H3cit levels and FXI activity. The association between PTS occurrence and 3 nominal predictors—presence in the top quartile of H3cit concentration (>3.775 ng/ml), top quartile of FXI activity (>112.15%), and intersection of H3cit and FXI top quartiles—was assessed using univariable logistic regression. Differences were considered significant if a 2‑sided P value was below 0.05. Statistical analyses were performed using IBM SPSS Statistics software, version 29 (IBM Corp., Armonk, New York, United States). Figures were prepared using the Matplotlib package for Python. Based on the previous study,12 the minimal detectable effect for FXI, with groups sizes of n = 20 and n = 152, is approximately 17.06%.
Results
We studied 95 men (55.2%) and 77 women (44.8%), at a median (IQR) age of 45 (33–55) years, including 128 (74.4%) with proximal DVT. In the whole group, median (IQR) FXI level was 102% (93%–112%), while the median (IQR) H3cit concentration was 2.9 (2.15–3.78) ng/ml. A positive correlation was observed between these variables (R = 0.359; P <0.001; Figure 1A). The patients with FXI activity equal to or above 120% (n = 24; 14%) had median (IQR) H3cit levels by 30% higher than those with FXI activity below 120% (3.73 [2.94–4.25] vs 2.87 [2.12–3.58] ng/ml; P = 0.006; Figure 1B).
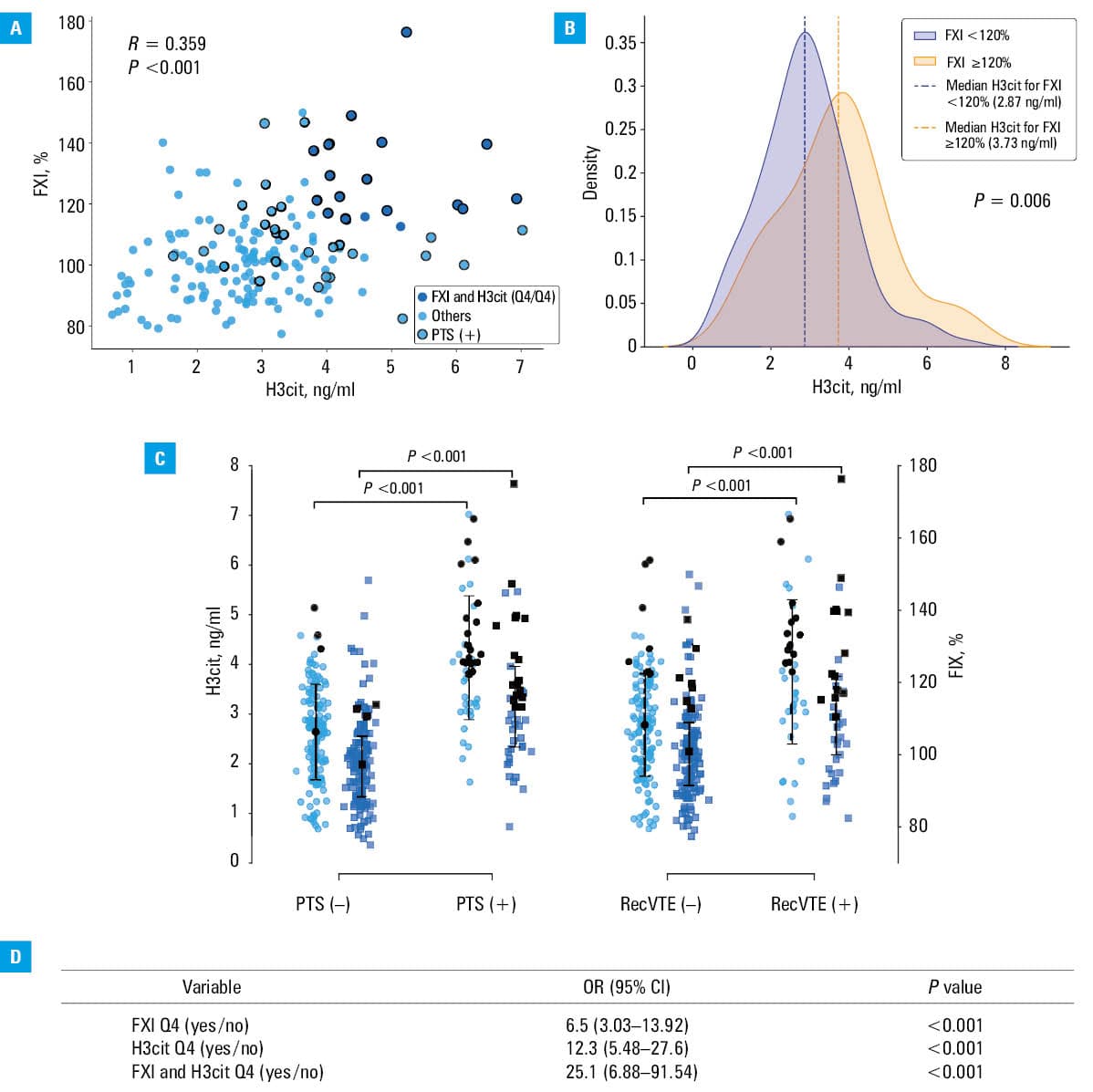
Abbreviations: FXI, factor XI; H3cit, citrullinated histone H3; OR, odds ratio; PTS(+), patients with post‑thrombotic syndrome; PTS(–), patients without post‑thrombotic syndrome; Q4, top quartile; recVTE(+), patients with recurrent venous thromboembolism; recVTE(–), patients without recurrent venous thromboembolism
At 1 year following DVT, PTS was diagnosed in 45 patients (26.2%), with a median (IQR) Villalta score of 15 (10.5–19.5). The patients with PTS had by 47.1% higher median (IQR) H3cit concentration than the remainder (4.03 [3.21–4.89] vs 2.74 [1.92–3.3] ng/ml; P <0.001; Figure 1C). Specifically, the patients with severe PTS (n = 24; 53.3%) had by 51.5% higher median (IQR) H3cit levels than those with milder forms (n = 9; 20%; 4.5 [4–5.59] vs 2.97 [2.38–3.28] ng/ml; P = 0.009). The median (IQR) FXI activity was by 15.8% higher in the patients with PTS than in those free of this disease (115.2% [104%–127.2%] vs 99.5% [90.6%–107.6%]; P <0.001; Figure 1C). FXI tended to be higher in severe PTS, as compared with mild PTS (118.8% [103.3%–137%] vs 105.9% [100.25%–111.7%]; P = 0.12).
VTE recurrence was observed in 41 patients (23.8%), who showed by 39.7% higher median (IQR) H3cit levels and by 9.3% higher median (IQR) FXI activities (3.87 [3.05–4.74] vs 2.77 [2.09–3.52] ng/ml and 110.5% [99.35%–122.05%] vs 100.8% [91.1%–109.3%], respectively; both P <0.001), as compared with those who did not experience such an event during median follow‑up of 53 (48–56) months (Figure 1C). The median duration of anticoagulant therapy was 10 (7–12) months. In all cases, temporary interruption or withdrawal of anticoagulation preceded VTE recurrence, after which medication was resumed in 38 patients (92.7%).
Twenty patients (11.6%) in the top quartile of both FXI activity (>112.15%) and H3cit level (>3.775 ng/ml) were older than the remainder (P <0.001) and more often had unprovoked VTE (P = 0.004) and proximal DVT (P = 0.03; Supplementary material, Table S1). In terms of inflammatory markers, CRP levels were by 43.1% higher in the top quartile than in the rest of the cohort. FXI activity was by 21.4% higher and H3cit concentrations were by 61.2% higher in the patients with the highest values of both markers (both P <0.001).
The patients in the top quartile for FXI activity alone had an OR for developing PTS of 6.5 (95% CI, 3.03–13.92), those in the top quartile for H3cit level had an OR of 12.3 (95% CI, 2.48–27.6), while the patients who were in the top quartile for both markers had an OR of 25.1 (95% CI, 6.88–91.54; Figure 1D).
The patients in the highest quartile for both markers developed PTS more frequently than the others (Supplementary material, Table S1). Specifically, the incidence of PTS in this subgroup was 4.62‑fold higher (17 [85%] vs 28 [18.4%]; P <0.001), and the median (IQR) Villalta score was 17 (12–20). Furthermore, at follow‑up, VTE recurred 3.53‑fold more frequently in the top quartile than in the rest of the cohort (13 [65%] vs 28 [18.4%]; P <0.001).
Discussion
To the best of our knowledge, this is the first study to report a positive correlation between plasma H3cit concentration and FXI activity in patients with DVT assessed 3 months after diagnosis. Previously, we found that elevated H3cit levels and higher FXI activity were each independently associated with the development of PTS.5,9 The current report extends previous evidence by showing that individuals with elevated FXI tend to exhibit more pronounced NETosis, detectable several months after VTE, despite ongoing anticoagulation, potentially contributing to DVT‑related complications during follow‑up.
Patients with FXI activity of 120% or greater had higher H3cit levels, suggesting a link between NETosis and FXI activity in thrombosis. NETs, through their DNA/histone scaffold, are considered to modulate blood coagulation by activating FXII, a primary component of the contact pathway, followed by FXI activation, promoting clot formation.4,13 In vitro studies have shown that NET‑associated proteins can promote clotting in FXI, FXII, and FVII‑deficient plasma, whereas neutrophils fail to induce coagulation in FXI‑deficient plasma.14 Moreover, recombinant human histones H3 and H4 exerted a procoagulant effect in platelet‑rich plasma, leading to thrombin generation. However, NETs themselves did not initiate the contact pathway or enhance thrombin‑dependent FXIa formation.15 Plasma FXI is strongly influenced by genetic determinants, with heritability estimated at approximately 45%. Genome‑wide association studies have identified common variants in the F11 gene and the KNG1 gene, which lead to constitutively elevated FXI activity throughout life, thereby contributing to a persistent prothrombotic state.16 In this study, elevated plasma FXI activity combined with higher H3cit concentrations were associated with an increased risk of PTS and recurrent VTE. This findings raise possibility that individuals with genetically elevated FXI activity may be susceptible to the prothrombotic effects of NETosis, particularly when NET formation is stimulated and reflected by increased amounts of circulating H3cit. Conversely, enhanced NETosis may amplify contact pathway activation through FXII‑mediated FXI activation, further increasing thrombotic risk.1 Of note, in the inferior vena cava stenosis murine model, FXI deficiency significantly reduced thrombus weight and incidence, and thrombi formed under these conditions had higher levels of H3cit.17 These observations suggested that NETs within thrombi occur independently of FXI, but their prothrombotic effects may amplify each other. Further mechanistic studies are needed to elucidate the molecular interplay between NETosis and elevated FXI, as the present study was not designed to address this issue.
It is worth mentioning that both reductions in the level of NET‑related proteins18 and inhibition of FXI19 exert anti‑inflammatory effects. This may indicate that interactions between NETs and the intrinsic coagulation pathway may have proinflammatory consequences that could contribute to the heightened risk of DVT complications.
Our study, which should be considered preliminary and hypothesis‑generating, has several limitations. First, the cohort was relatively small, although sufficient to detect significant intergroup differences. Second, H3cit level and FXI activity were measured at a single time point, precluding assessment of temporal variation. This may be relevant, as FXI levels may decrease when reassessed 3–6 months after VTE; however, baseline values appear to retain prognostic significance for PTS.9 Additionally, we did not measure FXIa or FXI‑inhibitor complexes to quantify active FXI, as well as additional circulating NET‑related proteins beyond H3cit, which could have provided a more comprehensive characterization of NETosis in DVT. Although H3cit is not entirely specific to NETosis, its biological stability, frequent use in clinical research, and potential for future standardization indicate its practical suitability as a biomarker, as compared with myeloperoxidase‑DNA complexes, which are characterized by limited specificity and a lack of standardized testing methods.5
In conclusion, this study demonstrates that circulating H3cit concentrations correlate positively with FXI activity, measured using a 1‑stage coagulometric assay, in patients with DVT. Moreover, our findings suggest that patients with both the highest level of H3cit and highest FXI activity constitute a subgroup at a particularly elevated risk of developing PTS and recurrent VTE.
- Noubouossie DF, Reeves BN, Strahl BD, Key NS. Neutrophils: back in the thrombosis spotlight. Blood. 2019; 133: 2186‑2197. | Crossref
- Natorska J, Ząbczyk M, Undas A. Neutrophil extracellular traps (NETs) in cardiovascular diseases: from molecular mechanisms to therapeutic interventions. Kardiol Pol. 2023; 81: 1205‑1216. | Crossref
- Urbanowicz T, Wojtasińska E, Olasińska‑Wiśniewska A, et al. Neutrophil to extracellular traps (NETs) as an early marker of right ventricular dilatation in patients with left ventricular assist devices (LVAD). Pol Heart J. 2024; 82: 777‑779. | Crossref
- Martinod K, Wagner DD. Reflections on targeting neutrophil extracellular traps in deep vein thrombosis. Arterioscler Thromb Vasc Biol. 2024; 44: 1719‑1724. | Crossref
- Krupa‑Zabiegała J, Michel PS, Stępień K, et al. Higher citrullinated histone H3 is associated with postthrombotic syndrome: a cohort study. J Thromb Haemost. 2025 Nov 3. [Epub ahead of print] | Crossref
SUPPLEMENTARY MATERIAL
ARTICLE INFORMATION