Diagnosis and management of pituitary neuroendocrine tumors: recent advances
1 Key words: multiomics, personalized medicine, PitNETs, pituitary neuroendocrine tumors, theragnosis
Key words: multiomics, personalized medicine, PitNETs, pituitary neuroendocrine tumors, theragnosis

 CC BY 4.0
CC BY 4.0
Diagnosis and management of pituitary neuroendocrine tumors: recent advances
Pituitary neuroendocrine tumors (PitNETs) are a heterogeneous group of neoplasms originating from anterior pituitary cells, encompassing a spectrum from indolent microadenomas to highly invasive, aggressive tumors. Their clinical presentation varies from classic endocrine hyper- or hyposecretory syndromes to mass effect symptoms or incidental imaging findings. The 2022 World Health Organization classification has redefined PitNETs, emphasizing their neuroendocrine nature and the central role of transcription factor–based immunohistochemistry. Recent years have witnessed rapid progress in molecular characterization, multiomics, radiomics, and theragnostic approaches, which are reshaping the approach to these tumors regarding their diagnosis, risk stratification, and management. This review provides a comprehensive and practical synthesis of current evidence, focusing on the integration of biomarkers, advanced imaging, and personalized therapies that could be useful in clinical practice, and discusses future directions in precision management of PitNETs.
Introduction
Pituitary neuroendocrine tumors (PitNETs), previously known as pituitary adenomas, are the most prevalent type of sellar neoplasms, accounting for 10%–15% of all intracranial tumors.1,2 While the majority are benign and slow‑growing, a clinically significant subset demonstrates invasive growth, recurrence, and resistance to conventional therapies. The 2022 World Health Organization (WHO) classification3 reconceptualized pituitary adenomas under the umbrella term “PitNETs,” recognizing the neuroendocrine biological origin, and emphasizing the importance of transcription factor profiling (pituitary‑specific transcription factor 1 [PIT1], steroidogenic factor 1 [SF‑1], TPIT [T‑box pituitary transcription factor]) for accurate lineage assignment and risk stratification.
Functioning PitNETs are characterized by hormone hypersecretion leading to distinct clinical syndromes: acromegaly (growth hormone [GH] excess), Cushing disease (adrenocorticotropic hormone [ACTH] excess), prolactinomas (prolactin [PRL] excess), and thyrotropinomas (thyroid‑stimulating hormone [TSH] excess). Nonfunctioning PitNETs (nfPitNETs), which constitute up to 35% of cases, typically present with symptoms of mass effect, or symptoms related to hormonal insufficiency due to the destruction of the pituitary parenchyma or they are frequently discovered incidentally.2
With improvements in molecular biology, imaging technology, and biomarker discovery, the management of PitNETs is shifting from a generalized to a personalized approach. This review highlights recent advances in diagnosis, prognosis, and treatment, with emphasis on theragnosis, multiomics, and radiomics. Special attention is given to guideline recommendations from the Endocrine Society, the European Society of Endocrinology (ESE), and the European Neuroendocrine Tumor Society.4-6
Methods
This narrative review encompasses literature published mostly between January 2018 and June 2025. A comprehensive search was performed in PubMed, Web of Science, and Scopus databases using the key words: “pituitary neuroendocrine tumors,” “PitNETs,” “theragnosis,” “personalized medicine,” “biomarkers,” “multiomics,” “radiomics,” and “targeted therapy.” Articles included were in English and comprised clinical guidelines, randomized controlled trials, systematic reviews, and translational research. Official position statements and consensus documents from the Endocrine Society and ESE were prioritized. References were supplemented by forward citation tracking and manual searching of reference lists from key publications. Data were selected for relevance to diagnosis, prognosis, treatment, molecular stratification, and imaging innovations in PitNETs.
Results
Biomarkers for diagnosis and prognosis
The diagnostic and prognostic assessment of PitNETs has evolved significantly in recent years, driven by advances in molecular pathology and the integration of multiomics approaches.
Functional PitNETs are characterized by the secretion of hormones, such as PRL, GH, ACTH, and TSH. Quantitative determination of circulating hormone levels by modern ultrasensitive assays is essential not only for diagnosis but also for follow‑up monitoring and early detection of recurrence.7 For example, random GH and insulin‑like growth factor 1 (IGF‑1) levels are typical of acromegaly, while 24‑hour urinary free cortisol and mid‑night salivary cortisol, along with ACTH determination, are diagnostic in ACTH‑secreting tumors. In contrast, nfPitNETs lack reliable circulating biomarkers so far, as α glycoprotein subunit has not been clinically validated, and are identified via imaging according to clinical compressive symptoms or symptoms of pituitary insufficiency and further postsurgically characterized in pathology studies.8
Immunohistochemistry (IHC) is now routinely combined with the assessment of transcription factors to assign tumor lineage as per the 2022 WHO classification.3,9 PIT1 lineage identifies tumors including somatotroph (GH‑secreting; further classified into densely and sparsely granulated variants), lactotroph (PRL‑secreting), and thyrotroph (TSH‑secreting) tumors, as well as several plurihormonal and less frequent subtypes, such as mammosomatotroph tumors (GH- and PRL‑cosecreting), acidophil stem cell tumors, immature PIT1‑lineage tumors (formerly referred to as silent subtype 3), and mature plurihormonal PIT1‑lineage tumors. TPIT presence marks TPIT‑lineage tumors, which are biologically corticotroph tumors. These can also be further subtyped into densely granulated, sparsely granulated, and aggressive Crooke cell tumors. SF‑1 lineage identifies primarily gonadotroph tumors (follicle‑stimulating hormone [FSH]- / luteinizing hormone [LH]-secreting), many of which are clinically nonfunctioning.9 Beyond these primary transcription factors, the classification also recommends the use of estrogen receptor α and GATA3 to refine the classification.3,10 Although transcription factors define lineage, hormone staining (eg, for ACTH, GH, PRL, βTSH, βFSH, βLH, and α-subunit) also remains essential to confirm hormone production and cellular differentiation.
Regarding acromegaly, Araujo et al11 studied GH- and PRL‑cosecreting PitNETs using data from a recent retrospective multicenter registry (ACRO‑SPAIN) that included more than 600 patients with acromegaly. Knosp grade above 2, elevated serum GH, and increased IGF‑1 levels were associated with surgical failure and resistance to first‑generation somatostatin receptor ligands (fgSRLs). A comprehensive review further highlighted that larger tumor size, cavernous sinus invasion, and specific GH/PRL staining patterns (characteristic of mixed subtypes) are often associated with worse surgical and medical outcomes, and higher recurrence risk.12
Additionally, cytokeratin patterns (eg, CAM 5.2) are important ancillary features for determining cell type and tumor subtype, such as the granulation pattern in somatotroph and corticotroph tumors, which, most importantly, correlates with aggressiveness and treatment response (eg, in somatotropinomas, sparsely granulated tumors correlate with reduced fgSRL treatment response, while densely granulated tumors correlate with better fgSRL treatment response).13 Prognostic IHC markers, such as Ki‑67/MIB1 index and p53 immunostains are also currently assessed in routine clinical practice.4 Ki‑67 is a marker of cell proliferation and is associated with more aggressive growth and poorer response to fgSRLs. While the Ki‑67 index above 3% was formerly used to define “atypical” adenomas, a denomination which has currently disappeared according to the new WHO classification,3 it now serves more broadly as a risk marker for risk stratification, as it is associated with increased risk of recurrence, lower responsiveness to medical treatment, and aggressive behavior, although this threshold must be interpreted in the clinical context of individual cases.3,14
Molecular biomarkers in PitNETs are an area of growing research interest, spanning genetic mutations, epigenetic alterations, and protein expression profiles; however, their clinical utility has not been fully established, and their study remains largely investigational. Among these, ubiquitin‑specific peptidase 8 (USP8) mutations in corticotroph tumors and methylguanine‑DNA methyltransferase (MGMT) expression in aggressive tumors have been most extensively studied and are discussed below.
Theragnosis and personalized medicine
The concept of theragnosis, where diagnostic findings are directly linked and concurrently guide therapeutic decisions, is being increasingly applied in the management of PitNETs.
One of the most significant advances in recent years is the use of somatostatin receptor (SSTR) profiling, particularly SSTR2 and SSTR5, to guide the selection of first- and second‑generation SRLs, such as octreotide and pasireotide, respectively.15,16 For somatotroph tumors, robust SSTR2 expression is a key predictor of favorable biochemical response to fgSRLs, such as octreotide and lanreotide, and the opposite, its low expression, is even more strongly supporting a poor response to these therapeutic compounds.15,16 Complementing SSTR profiling, Coopmans et al17 identified baseline IGF‑1 concentration as the best clinical predictor of biochemical response to fgSRLs in acromegaly. Other key factors included body weight and age, with higher body weight and younger age being associated with poorer response rates. In contrast, pasireotide, a second‑generation SRL, exhibits high binding affinity for multiple SSTRs, notably SSTR5 and SSTR2 (SST5 > SST2). Its efficacy in corticotroph tumors is predominantly linked to high expression of SSTR5 in these tumors, although the presence of SSTR2 seems to be a permissive condition. However, in GH‑omas that are resistant to fgSRLs and show negligible SSTR2 expression, pasireotide may still exert effects through activation of SSTR5 alone.15 SSTR3 has been consistently implicated in inducing apoptosis across multiple cell types, as shown in early and more recent mechanistic studies expressing SSTR3 in vitro and demonstrating p53‑mediated apoptosis.18 More recently, Di Muro et al19 demonstrated that a full SSTR3 agonist, ITF2984, exerted antimitotic and proapoptotic effects on human nfPitNET cells. Pasireotide, as a somatostatin multireceptor ligand, exhibits moderate binding to SSTR3; however, no specific evidence has currently demonstrated that pasireotide causes tumor shrinkage in PitNETs via SSTR3‑mediated apoptosis. Thus, while SSTR3 is a biologically plausible and functionally validated target associated with apoptosis and tumor growth inhibition, the exact mechanism of pasireotide action likely involves a broader interplay of receptor subtypes. Despite these insights, the direct correlation and interplay of receptor expression with drug efficacy are not always straightforward, and call for standardized validation methods for SSTR expression in routine clinical practice persist.15,16
E‑cadherin has also emerged as a particularly strong and practical biomarker for predicting therapeutic response in acromegaly.20,21 The study by Puig‑Domingo et al16 provides substantial evidence for its superior predictive power over SSTR2, suggesting its clinical utility in guiding treatment decisions. The ACROFAST (Personalized Medicine in Acromegaly) study further demonstrates the successful integration of E‑cadherin into a personalized treatment algorithm, confirming its feasibility and predictive value in a real‑world clinical setting.22 Therapeutic decisions in acromegaly are an evolving example of precision medicine in PitNETs, and should incorporate not only biochemical activity, but also tumor size, consistency, invasiveness, and molecular expression profiles.16 Recent evidence supports the use of multiparameter approaches that integrate clinical, imaging, and molecular data to guide individualized treatment strategies.22
In lactotroph tumors, dopamine receptors play a pivotal role, where the presence of D2 dopamine receptors predicts responsiveness to dopamine agonists (DAs), such as cabergoline.2,23 DAs are also occasionally used off‑label in GH‑secreting PitNETs expressing D2 receptors.23 Hotspot somatic mutations in splicing factor 3 subunit B1 (SF3B1R625H) have been detected in 20% of prolactinomas. These cases show higher PRL levels and shorter progression‑free survival, as compared with patients without the mutation. This mutation causes aberrant splicing of estrogen‑related receptor γ, which results in stronger binding of PIT1, leading to excessive PRL secretion.24
For aggressive PitNETs, MGMT expression, which determines suitability for alkylating chemotherapy, has been associated with a better response to temozolamide (TMZ) treatment.5,25 However, it should be noted that the latest version of the ESE guideline on aggressive pituitary tumors no longer suggests routine MGMT IHC before trialing TMZ, as MGMT protein IHC has not been fully validated to predict TMZ response, which differs from the previous recommendation.4,6 These aggressive tumors should be followed at short term (3–6‑month interval) and metastatic disease should be investigated, mostly with imaging procedures. A multidisciplinary management strategy is essential to ensure coordinated diagnostic assessment and individualized therapeutic planning for all pituitary tumors, but particularly for these highly aggressive ones.
Functional imaging has significantly advanced theragnostic strategies in the management of PitNETs, enabling more personalized and targeted approaches to patient care. The use of 68Ga‑DOTA‑0‑Tyr3‑Octreotate in positron emission tomography / computed tomography (DOTATATE PET/CT), which binds with high affinity to SSTR2, allows for precise localization of SSTR2‑expressing tumors and plays a critical role in identifying candidates for peptide receptor radionuclide therapy (PRRT), typically using lutetium‑177‑labeled somatostatin analogues.26,27 PRRT is currently under investigation as a therapeutic option for aggressive, recurrent, or metastatic PitNETs that express SSTR2, particularly in patients who have not responded to conventional treatments, such as TMZ.26
Targeted therapies and emerging strategies
The management of PitNETs is evolving from a one‑size‑fits‑all approach to a more stratified, subtype‑specific strategy, supported by advances in molecular characterization and the development of targeted therapies. Medical treatment is increasingly being tailored according to tumor subtype, hormonal secretion profile, and underlying molecular alterations.
Prolactinomas are effectively managed with DAs as the first‑line therapy in most patients.28 Cabergoline, a long‑acting oral DA, is highly effective at lowering serum PRL levels and reducing tumor size in 50%–70% of prolactinomas. It is generally more effective and better tolerated than bromocriptine. DAs are the first‑line therapy for all prolactinoma types, including giant prolactinomas, while transsphenoidal surgery (TTS) is reserved for DA‑resistant cases (about 10% or less), when more than 3 mg/week are required or when treatment fails to reduce tumor size by over 50% at maximum tolerable doses,29 or for tumors presenting with acute mass effects, such as apoplexy or rapid / progressive vision loss.23,28 Additionally, careful follow‑up is required in giant prolactinomas that may present a very rapid tumor shrinkage response to DAs causing cerebrospinal fluid fistula, usually requiring surgical repair.30
While TTS remains the first‑line treatment for acromegaly, fgSRLs, such as octreotide and lanreotide, have been until now the standard medical options when surgery is not feasible31 or to complement insufficient surgical resection. In patients with resistant or partially controlled disease, GH receptor antagonist pegvisomant or second‑generation SRL pasireotide may be considered, based on individual comorbidities and tolerability. Combination therapy with SRLs and either pegvisomant or cabergoline could be appropriate in suboptimal responders, but the challenge is to predict which one is the most appropriate treatment for a given case. Radiotherapy is reserved for refractory cases due to its long‑term side effect profile.31 Recent studies support personalized treatment strategies in acromegaly to achieve hormonal control more rapidly. The ACROFAST study22 explored the effectiveness of tailoring initial medical therapy (using fgSRLs, pegvisomant, or combination regimens) based on predictive biomarkers, including the short acute octreotide test (sAOT), T2‑weighted magnetic resonance imaging (MRI) signal, and E‑cadherin expression. A significantly higher proportion of patients following this personalized protocol vs the standard approach (which consistently initiated treatment with fgSRLs) achieved hormonal control (78% vs 53%; P <0.05) over a 12‑month follow‑up period. This personalized strategy also resulted in control being achieved in a shorter period of time (median of 182 d) than for the standard treatment (median of 305 d).22
Corticotroph tumors not responding to TTS remain particularly challenging. Medical tumor‑directed treatment to control hypercortisolism includes pasireotide, long‑acting‑release pasireotide, and cabergoline. Pasireotide shows efficacy in 20%–40% of patients despite potential hyperglycemia and potential escape phenomenon at mid‑term follow‑up. Steroidogenesis inhibitors (eg, ketoconazole, levoketoconazole, metyrapone, osilodrostat, mitotane, etomidate) can control cortisol levels without directly addressing the pituitary tumor. Radiotherapy remains a slower‑acting alternative for disease control and is commonly used alongside medical therapy.32
USP8 mutations are frequently identified in ACTH‑secreting PitNETs, occurring in 21%–62% of cases and being specific to this tumor type.33 These mutations enhance epidermal growth factor receptor (EGFR) signaling by preventing proteasomal degradation increasing ACTH production. Although USP8‑mutant tumors are often small and initially achieve surgical remission, some studies suggest a higher recurrence risk, and their overall prognostic value remains unclear; nevertheless, the identification of USP8 mutations has advanced understanding of corticotroph tumor biology and indicates a potential, yet still investigational, role for EGFR‑targeted therapies.33 Expression profiling of USP8‑mutant vs wildtype corticotroph PitNETs revealed over 1600 differentially expressed genes, affecting cell signaling, adhesion (CDH6, GJA1), ion transport (KCNN4), γ-aminobutyric acid signaling, and cell cycle regulation (CDC25A, CDK6). These findings underscore the pleiotropic impact of USP8 mutations beyond EGFR regulation.34
For aggressive and treatment‑refractory PitNETs, TMZ remains the only validated chemotherapy, and it is recommended as the first‑line treatment by the 2025 Revised ESE Clinical Practice Guideline for aggressive pituitary tumors and pituitary carcinomas.4 TMZ monotherapy is advised following documented progression after other multimodal treatments, although the level of evidence remains low. Treatment should be continued for 12 months in responding patients, while durations beyond 24 months are discouraged due to the risk of cumulative toxicity.4-6 Despite achieving an objective radiological response in about 37% of cases, most patients ultimately experience progression. In such scenarios, immune checkpoint inhibitors (ICIs) are now specifically suggested for pituitary carcinomas with rapid progression after TMZ failure, supported by data showing efficacy in tumors with mismatch repair deficiency or high tumor mutational burden.35 Bevacizumab (a vascular endothelial growth factor inhibitor) is considered experimental and may be used only when ICIs are not accessible.6 PRRT using radiolabeled somatostatin analogues, such as 68Ga‑DOTATATE for diagnostic imaging, is a new possible strategy for SSTR‑positive, treatment‑resistant PitNETs,26 but it remains an investigational option for highly selected SSTR‑positive tumors with high ligand uptake. Other experimental therapies under investigation include mammalian target of rapamycin inhibitors (such as everolimus)36 and EGFR inhibitors (such as lapatinib),37 but current clinical evidence is sparse and primarily limited to preclinical or early‑phase studies. The guidelines emphasize the need for molecular testing in progressing tumors to identify targetable alterations and support enrollment in clinical trials exploring these emerging therapies.4 Table 1 summarizes the status of systemic therapies for aggressive pituitary tumors and pituitary carcinomas according to the 2025 Revised ESE Clinical Practice Guideline.4 A recent observational study by Bioletto et al38 evaluated 123 patients with PitNETs treated with radiotherapy, with a median follow‑up of 10 years. Tumor progression after radiotherapy was observed in 23% of cases. The study identified lactotroph and corticotroph subtypes, male sex, and necrotic‑hemorrhagic changes on pretreatment MRI as independent risk factors for progression. The patients experiencing progression frequently required additional treatments, including surgery, TMZ, or reirradiation. These findings highlight that, although radiotherapy controls tumor growth in most cases, a subset of PitNETs remains at a high risk for recurrence, underscoring the need for careful long‑term follow‑up and individualized management strategies, ideally within a multidisciplinary framework.
Therapeutic agent | Treatment | Radiological response rate | ESE recommendation (strength / evidence quality) | Clinical indication | Evidence source11 |
Abbreviations: CR, complete response; EGFR, epidermal growth factor receptor; ESE, European Society of Endocrinology; ICI, immune checkpoint inhibitor; MMRd, mismatch repair deficiency; mTOR, mammalian target of rapamycin; PC, pituitary carcinoma; PR, partial response; PRRT, peptide receptor radionuclide therapy; TMB, tumor mutational burden; TMZ, temozolomide; SSTR, somatostatin receptor; VEGF, vascular endothelial growth factor | |||||
TMZ | Cytotoxic chemotherapy | 37% (CR + PR) | Recommended (strong) / low‑quality evidence | First‑line; continue treatment for 12 months in responding patients; evaluate response after 3 cycles | Meta‑analysis of 11 studies and 4 surveys (n = 439) |
ICIs | Immunotherapy: pembrolizumab, nivolumab, ipilimumab | 24% PR | Suggested (weak) / investigational treatment | Second‑line; PC patients with rapid progression, especially if molecular testing shows MMRd or high TMB | Synthesis of 3 cohort studies (n = 25) |
Bevacizumab | VEGF inhibition (antiangiogenesis) | Minimal (1/11 PR in surveyed cohorts) | Experimental / limited option | Consider if ICI access is restricted | Second ESE survey (n = 11) |
PRRT | Radioligand therapy (SSTR‑targeting) | Low (4/19 patients achieved PR) | Investigational treatment | Selected tumors that are SSTR‑positive with high ligand uptake | Case reports and small series (n = 19) |
Other targeted agents (everolimus, lapatinib) | mTOR / EGFR inhibition | Sparse clinical data; anecdotal responses only | Experimental | Use is generally discouraged outside clinical trials | ESE surveys and case reports |
Double PitNETs, defined as 2 distinct tumors coexisting in the same gland, remain a challenging clinical setting requiring multidisciplinary expert care,39,40 particularly when affecting the pituitary gland. This complexity is stressed by variants, such as asynchronous double adenomas, which may initially present as a classic single‑tumor phenotype. While MRI is the primary diagnostic tool, it often complicates diagnosis, as nearly half of double PitNETs (48%) appear radiologically as a single, contiguous tumor rather than 2 distinct lesions (45%), obscuring the second component.40 To overcome this limitation, particularly in diagnosing small ACTH‑producing lesions, advanced radiological strategies are suggested, including the use of thin‑slice imaging with 3 Tesla (3T) MRI, thorough examination using spoiled gradient‑recalled acquisition sequences, and the utilization of methionine‑PET fusion 3T‑MRI.32 Ultimately, a definitive diagnosis relies on detailed pathological examination, which has been refined by the 2022 WHO classification.3 Modern pathology integrates hormonal staining with the assessment of pituitary transcription factors (PIT1, TPIT, and SF1). This approach is crucial for distinguishing double PitNETs from the heterogeneity of a single lesion, especially when the tumors are adjacent but exhibit different transcriptional lineages. Despite these diagnostic advancements, clinicians and pituitary neurosurgeons must maintain high awareness of hidden microtumors during surgical exploration, as noninvasive imaging often fails to detect both lesions preoperatively.
Multiomics in tumor stratification
Multiomics technologies, including genomics, transcriptomics, epigenomics, and proteomics, are redefining the classification and prognostication of PitNETs, although their clinical application remains largely investigational.
Genomic studies have shown that GNAS mutations are present in approximately one‑third of somatotroph PitNETs, where they cause constitutive activation of the cyclic adenosine monophosphate pathway, driving excess GH secretion. However, the presence of these mutations does not significantly influence clinical aggressiveness or the response to SRL therapy.41 In contrast, USP8 mutations are commonly found and are specific to ACTH- secreting PitNETs.33,41 While they do not currently alter clinical management, their identification helps refine molecular classification and may guide future targeted therapies.
Transcriptomic analysis, utilizing whole‑exome sequencing, identifies distinct genomic profiles across PitNETs. A study by Andonegui‑Elguera et al42 demonstrated more genomic alterations in ACTH‑secreting lesions causing Cushing disease than in silent corticotroph PitNETs, with ACTH‑secreting carcinomas exhibiting the highest abnormalities, underscoring their aggressive biological behavior and suggesting an evolutionary continuum.
Epigenomics has emerged as a powerful tool for distinguishing aggressive / metastatic PitNETs from benign ones. Genome‑wide methylation profiling reveals distinct epigenetic signatures and frequent copy‑number variations in aggressive tumors, even in initial surgical samples. These features are typically absent in benign PitNETs. Such molecular differences offer early prognostic insight. This supports the potential for intensified surveillance and earlier adjuvant therapy, despite benign histological appearance.43,44
Proteomics is getting increasing importance for characterizing PitNET heterogeneity, reflecting the functional state and coding transcriptome. Quantitative proteomic analyses reveal distinct signatures correlated with clinical behavior, often drawing upon extensive protein databases. Key findings related to aggressiveness include the upregulation of solute carrier family 2 member 1 associated with invasiveness and epithelial‑to‑mesenchymal transition, as identified by Zhang et al45 through quantitative proteomics comparing invasive and noninvasive pituitary adenomas. Similarly, in a study by Chen et al,46 high expression of cathepsin Z was found in invasive somatotroph PitNETs. Functional profiling via post‑translational modifications is also critical; for instance, phosphorylation of β-catenin at serine 552 was found to be related to the invasion and recurrence of nfPitNETs.47 Ultimately, integrated proteogenomic approaches enable refined classification by defining distinct molecular clusters based on proteomic profiles, which is essential for biomarker discovery, risk stratification, and guiding precision medicine strategies.23,33,48
Advances in imaging and radiomics
MRI remains a cornerstone for the diagnosis and planning surgical removal of PitNETs, offering crucial anatomical insights. However, traditional visual assessment of MRI scans offers limited information regarding the method’s ability to fully capture the complex biological properties of these heterogeneous tumors, while radiomics addresses this limitation by transforming conventional MRI scans into high‑dimensional, mineable quantitative data, allowing for the prediction of tumor consistency, cavernous sinus invasion, Knosp grade, postoperative recurrence, and even histological subtype. Radiomic signatures have proven highly informative for characterizing granular patterns and predicting responses to pharmacologic treatment in acromegaly. Notably, Park et al49 developed a radiomics model based on T2‑weighted MRI that distinguished densely granulated from sparsely granulated GH‑secreting PitNETs in 69 acromegaly patients. The model built, using 4 selected radiomic features and validated via LASSO logistic regression with 5‑fold crossvalidation, achieved an area under the receiver operating characteristic curve (AUC) of 0.834 (95% CI, 0.738–0.93), significantly outperforming qualitative T2 assessments (AUC, 0.597) and relative signal intensity evaluations (AUC, 0.647).49 Furthermore, Fan et al50 employed a multiparametric MRI radiomics analysis combined with machine learning (support vector machine plus clinical features), showing that the radiomics signature could noninvasively predict radiotherapeutic response in acromegaly patients. These features can be integrated into predictive models to guide surgical planning and risk stratification, representing a significant step toward precision imaging in PitNETs.51,52 Radiomics and PET/CT fusion imaging are especially valuable in preoperative cases and in tumors with atypical or aggressive biological behavior, where conventional imaging may be limited.27,51
As previously mentioned, functional imaging modalities, such as 68Ga‑DOTATATE PET/CT, have emerged as valuable tools for the detection of SSTR expression, particularly in aggressive or recurrent tumors, and selecting candidates for PRRT.26
In addition, ¹¹C‑methionine PET/CT is employed in selected centers for the detection of residual or recurrent pituitary lesions, particularly when findings on conventional MRI are inconclusive.53
The integration of radiomics with clinical, pathological, and molecular data within a multidisciplinary framework is paving the way for the development of comprehensive predictive algorithms and personalized management strategies in PitNETs, as shown in Figure 1.
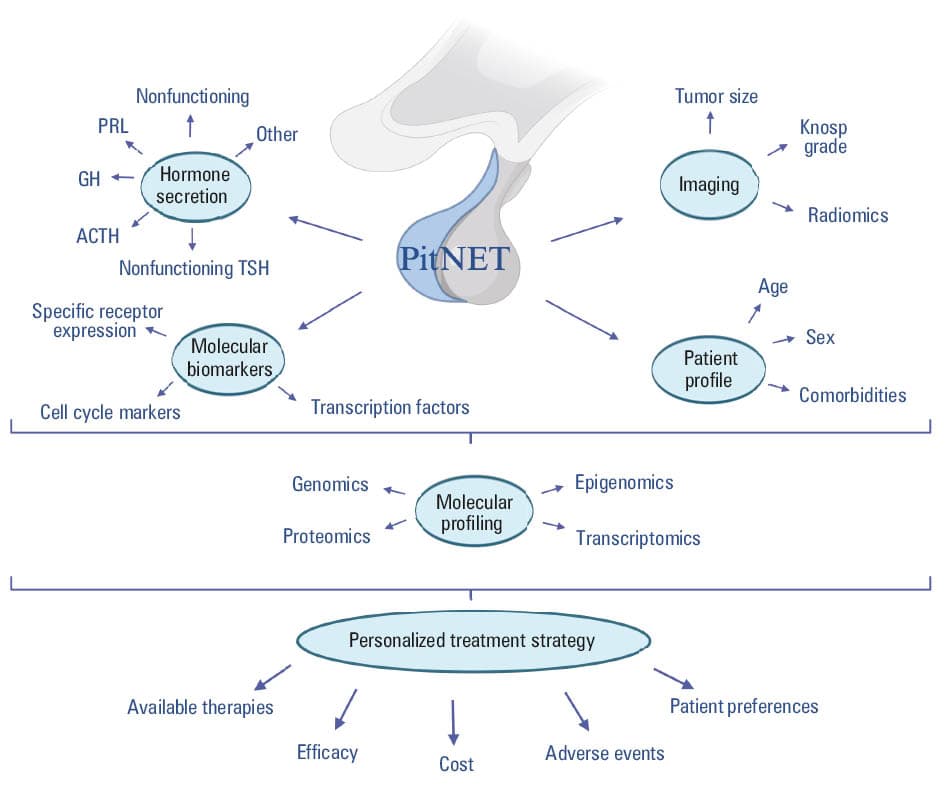
Abbreviations: ACTH, adrenocorticotropic hormone; GH, growth hormone; PitNET, pituitary neuroendocrine tumor; PRL, prolactin; TSH, thyroid‑stimulating hormone
Functional testing and metabolomics
The sAOT has recently emerged as a potentially valuable tool for predicting treatment responses in acromegaly, offering a dynamic, clinic‑friendly alternative to other more time‑consuming functional assays.54 In a post hoc study from the ACROFAST trial,54 a GH cutoff of 1.4 ng/ml at 2 hours after a 100 µg subcutaneous somatostatin injection achieved 81% accuracy, 73.3% sensitivity, and 94.1% specificity in distinguishing responders to fgSRLs from nonresponders, with the former having significantly lower GH levels at 2 hours. Notably, tumor expression of E‑cadherin and SSTR2 correlated with GH level at 2 hours, validating the predictive utility of sAOT in personalizing SRL therapy.
In parallel, metabolomics is paving a transformative path toward functional phenotyping of PitNETs. Through both liquid chromatography–mass spectrometry and nuclear magnetic resonance (NMR)-based approaches, researchers have characterized distinct metabolic signatures across PitNET subtypes. Pînzariu et al55 systematically reviewed PitNET metabolomics, noting altered amino acid metabolism—including downregulation of phosphoethanolamine, N‑acetyl aspartate, and myoinositol in prolactinomas, and increases in aspartate, glutamate, and glutamine in functioning tumors. In Cushing disease, metabolomic markers, such as deoxycholic acid, 4‑pyridoxic acid, and glucose‑6‑phosphate emerged as promising biomarkers. Moreover, intraoperative matrix‑assisted laser desorption / ionization mass spectrometry imaging demonstrated potential in distinguishing functional PitNETs from healthy pituitary tissue to assist surgical planning.55
Complementing these findings, Ijare et al56 applied ex vivo 1H‑NMR spectroscopy to resected PitNET specimens across hormone subtypes. Their profiling revealed subtype‑specific metabolite trends, for example, decreased levels of N‑acetylaspartate, myo- and scyllo‑inositol, glycine, taurine, and phosphoethanolamine in prolactinomas; increased levels of aspartate in ACTH‑secreting tumors; and reduced level of glycerophosphoethanolamine across PRL‑secreting, ACTH‑secreting, and nfPitNETs, highlighting distinct metabolic fingerprints that could guide functional subclassification.56
Further broad‑based multiomic integration by Feng et al57 demonstrated that PitNETs exhibit downregulated glucose metabolism and glycolysis, alongside altered amino acid and fatty acid pathways. Through a combination of metabolomic, transcriptomic, and proteomic analyses across 8 PitNET subtypes, their study identified isocitrate dehydrogenase (IDH2) as a key metabolic player in GH‑secreting tumors, with functional implications in regulating tumor growth and hormone secretion. Notably, inhibiting IDH2 impaired tumor cell proliferation in vitro, suggesting a potential metabolic therapy target.57
Together, these innovative methodologies—sAOT and metabolomics—offer a compelling route to refine functional testing in PitNETs. The sAOT provides a rapid, predictive assessment of fgSRL responsiveness linked to tumor‑specific biomarkers (SSTR2 and E‑cadherin), enabling tailored pharmacologic planning.54 Metabolomic profiling, by contrast, captures the final biochemical phenotype of tumors, offering a nonhormonal, molecularly informed lens for classification, prognosis, and intraoperative guidance.55,57 Unlike conventional tests that measure hormone levels only, metabolite profiling can uncover subtype‑specific metabolic imprints and signal actionable pathways (eg, IDH2), thus elucidating the mechanisms of tumor behavior and response potential.
While some of these tools—particularly metabolomic profiling—remain in early development and require broader validation, they illustrate a shift toward functionally and molecularly informed diagnostics. Future studies should aim to standardize metabolomic panels, validate sAOT thresholds across diverse populations, and integrate these data with imaging and molecular markers. Ultimately, combining dynamic functional tests, such as sAOT, with metabolomic phenotyping could supplant or enhance traditional functional assays, paving the way for precision medicine in PitNET care.
Discussion
The diagnostic and therapeutic landscape of PitNETs is undergoing a paradigm shift. Historically considered benign and classified solely based on hormone IHC, PitNETs are now appreciated as complex neuroendocrine tumors requiring nuanced classification and personalized treatment. Advances in IHC profiling, particularly with transcription factors such as PIT1, TPIT, and SF‑1, have provided the basis for improved subtyping and risk stratification.3,13
The implementation of theragnostic approaches, including SSTR‑based imaging and receptor‑targeted therapies, marks an important leap toward individualized care, especially in aggressive or recurrent PitNETs. Molecular profiling and functional PET imaging (eg, 68Ga‑DOTATATE) now complement conventional MRI in guiding systemic treatments such as TMZ, ICIs, or PRRT.4-6,25,26
Multiomics integration, encompassing genomics, transcriptomics, proteomics, and epigenomics, offers further granularity for tumor stratification. The clinical relevance of GNAS and USP8 mutations in functioning PitNETs is well established, and ongoing efforts to map the methylome and proteome may soon deliver novel prognostic and predictive biomarkers.33,41,44,48
Our group has consistently highlighted the variability of acromegaly presentation and treatment response, underlining the need for tailored management strategies based on tumor subtype, invasiveness, pharmacogenomics, and imaging phenotypes. Personalized treatment in PitNETs is now a reality, supported by predictive models and clinical decision frameworks.22
Emerging techniques in radiomics are already influencing clinical decision‑making, with algorithms capable of predicting cavernous sinus invasion, response to radiosurgery, and even histological subtype based solely on MRI features.51,52 Despite these advances, significant challenges remain. There is a need for prospective validation of multiomics and radiomics biomarkers across diverse populations, as well as for the development of standardized, widely accessible diagnostic algorithms. The translation of emerging therapies, such as ICIs and novel targeted agents, into routine clinical practice will require robust clinical trial evidence and international collaboration. In addition, optimal management of PitNETs requires a multidisciplinary approach involving endocrinologists, neurosurgeons, pathologists, radiologists, and oncologists. Such coordinated collaboration is essential to ensure accurate diagnosis, effective therapeutic decision‑making, and long‑term follow‑up.
Implementing the theragnostic concept in PitNETs medicine drives us toward a predictive framework scenario, which will definitely integrate multidimensional data in PitNETs biology. The journey to classify PitNETs reflects a growing understanding of their profound biological heterogeneity. Initial systems, grounded in histology and immunohistochemistry, provided a foundational taxonomy but proved inadequate for predicting clinical behavior, as tumors of identical morphological appearance could diverge significantly in their aggressiveness and recurrence potential. The subsequent revisions by the WHO in 2017 and 2022,3 which incorporated transcription factors and key molecular markers, represented a critical evolution toward a more biologically relevant classification.3 This shift acknowledged that tumor lineage, while informative, is only one piece of a complex puzzle. Despite these advances, a significant translational gap persists. Contemporary WHO criteria3 offer improved diagnostic accuracy but remain largely descriptive, failing to reliably stratify risk, predict therapeutic response, or guide personalized management strategies. Clinicians are thus often left with diagnostic certainty but prognostic uncertainty, unable to answer fundamental questions about a tumor’s potential for invasion or recurrence based on a pathological report alone. This limitation is starkly evident in the variable clinical courses of tumors within the same molecular subtype and in the frequent delays in diagnosing complex endocrine syndromes, where a static pathological classification offers little proactive insight.
This gap underscores an urgent need for a new generation of classification systems that are not merely descriptive but also predictive. The emergence of high‑throughput multiomics technologies, namely genomics, epigenomics, transcriptomics, and proteomics, has begun to unravel the molecular drivers of this heterogeneity, revealing a landscape far more complex than previously appreciated. However, the sheer volume and complexity of these data present a formidable challenge: how can these multidimensional datasets be synthesized into clinically actionable knowledge?
Therefore, the future of PitNET management lies in the development of integrative analytical frameworks. Such frameworks are required to harmonize traditional histopathological features with the deep molecular layers provided by omics technologies and rich clinical phenotyping. The goal is to move beyond categorical labels and toward continuous, biologically informed models that can quantify risk and predict behavior. This endeavor is not without its challenges; it is hampered by the scarcity of large, well‑annotated patient cohorts and the inherent variability in data collection across institutions. Initiatives such as the European Registries for Cushing syndrome (ERCUSYN) and REMAH (Spanish pituitary tumor registry) highlight the field’s recognition that overcoming these obstacles requires large‑scale, multi‑institutional collaboration to ensure robust and generalizable findings.
Ultimately, the application of sophisticated computational tools is indispensable for this task. These advanced analytical techniques are uniquely capable of identifying subtle, nonlinear patterns across disparate data types, pinpointing correlations between molecular signatures and clinical outcomes that elude conventional statistics. Crucially, for clinical adoption, the focus must extend beyond raw predictive power to include model interpretability, ensuring that the biological and clinical drivers of a prediction are transparent to the physician. The convergence of comprehensive data integration and powerful computational analysis has the potential to transform PitNET care, paving the way for a future where diagnostic reports not only categorize a tumor but also forecast its behavior and inform precise, personalized therapeutic interventions.
Conclusions
In conclusion, the future of PitNET management lies in precision endocrinology: combining histopathological advances, molecular profiling, functional imaging, and machine‑learning–powered diagnostics to optimize outcomes for individual patients. These innovations enhance diagnostic accuracy, refine prognostic stratification, and broaden therapeutic options, particularly for aggressive or treatment‑resistant tumors. Continued research and multidisciplinary collaboration will be key to translating these advances into routine clinical practice and improving outcomes for patients with PitNETs.
- Albano L, Losa M, Barzaghi LR, Mortini P. Benign and malignant tumors of the pituitary gland. In: Rezaei N, Hanaei S, eds. Human Brain and Spinal Cord Tumors: From Bench to Bedside. Volume 2. Cham: Springer International Publishing, 2023: 281‑297. | Crossref
- Tritos NA, Miller KK. Diagnosis and management of pituitary adenomas: a review. JAMA. 2023; 329: 1386. | Crossref
- Asa SL, Mete O, Perry A, Osamura RY. Overview of the 2022 WHO classification of pituitary tumors. Endocr Pathol. 2022; 33: 6‑26. | Crossref
- Raverot G, Burman P, Abreu AP, et al. Revised European Society of Endocrinology Clinical Practice Guideline for the management of aggressive pituitary tumours and pituitary carcinomas. Eur J Endocrinol. 2025; 192: R45‑R78. | Crossref
- McCormack A, Dekkers OM, Petersenn S, et al. Treatment of aggressive pituitary tumours and carcinomas: results of a European Society of Endocrinology (ESE) survey 2016. Eur J Endocrinol. 2018; 178: 265‑276. | Crossref
ARTICLE INFORMATION