Primary care provider–initiated screening for familial hypercholesterolemia: a model proposed based on the Kordian primary prevention program for cardiovascular diseases
Key words: Dutch Lipid Clinic Network scale, familial hypercholesterolemia, genetic testing, primary cardiovascular prevention

 CC BY 4.0
CC BY 4.0
Primary care provider–initiated screening for familial hypercholesterolemia: a model proposed based on the Kordian primary prevention program for cardiovascular diseases
Introduction: Familial hypercholesterolemia (FH) is a group of monogenic disorders causing high low‑density lipoprotein cholesterol (LDL‑C) levels and early cardiovascular diseases (CVDs).
Objectives: We aimed to present a primary care provider (PCP)-initiated FH screening model built at the University Hospital in Kraków as part of the Kordian CVD prevention program.
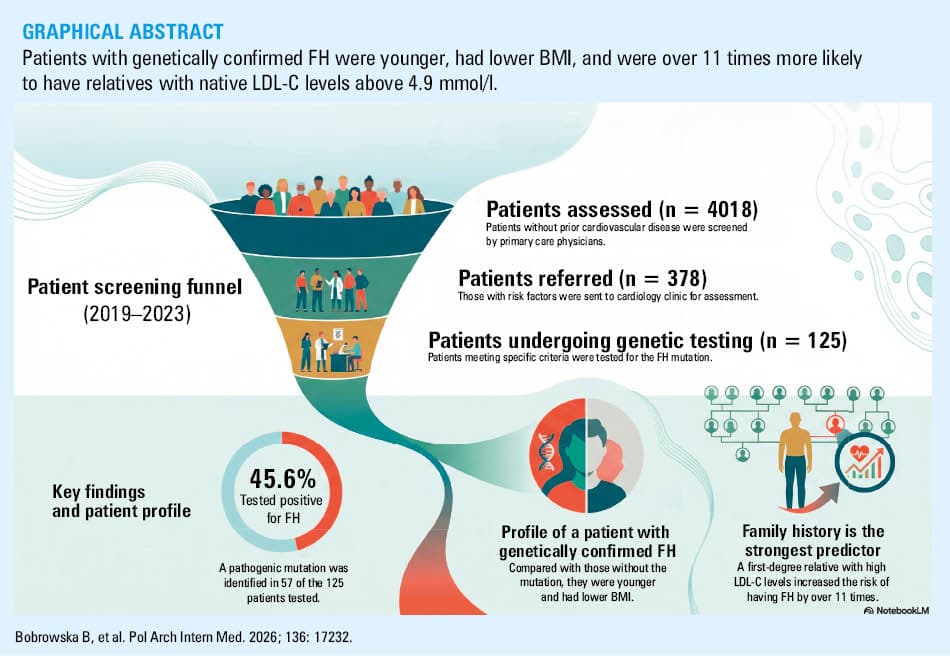
Patients and methods: Patients without a prior diagnosis of any chronic disease, including CVD, and who had not taken any medications, were selected by PCPs as eligible to participate. The study, conducted from 2019 to 2023, involved 4018 patients. From this group, 378 individuals (9.4% of the study population) with CVD risk factors were referred by PCPs to the Department of Cardiology for further assessment.
Results: Overall, 125 patients (33.1% of those referred to the Department of Cardiology), including 51 men and 74 women, met the clinical criteria and underwent genetic testing for FH. Their mean (SD) age was 46.2 (13.1) years. The individuals with confirmed pathogenic heterozygous FH mutations accounted for 45.6% (n = 57) of the population selected for genetic testing. The patients with genetically confirmed FH were notably younger (median [interquartile range], 41 [34–52] vs 49 [39.25–61.75] y; P = 0.002), and had lower body mass index (mean [SD], 24.59 [3.61] vs 27.22 [3.31] kg/m2; P = 0.009) than those without a genetic diagnosis. Twice as many patients with confirmed FH had relatives with native LDL‑C levels exceeding 4.9 mmol/l (71.9% vs 27.9%; P <0.001). This factor increased the risk of FH by more than 11 times.
Conclusions: Collaboration between PCPs and specialists with access to genetic testing, guided by a well‑designed algorithm, may facilitate the effective identification of patients with FH mutations before CVD develops.
What's new?
Genetic testing for familial hypercholesterolemia (FH) in Poland is typically performed as part of secondary prevention after a young person has experienced a cardiovascular event and a significantly elevated low‑density lipoprotein cholesterol level has been detected. Here, we propose a model for initiating FH testing by primary care providers (PCPs) as a primary cardiovascular prevention. The Polish Ministry of Health funded the Kordian program at the University Hospital in Kraków, enabling screening for cardiovascular risk factors in over 4000 participants without prior diagnosis. The protocol permitted genetic testing for the individuals who met the clinical criteria of FH. A total of 125 patients were referred for FH genetic testing. We identified 57 individuals with a pathogenic FH mutation. This report has direct clinical implications, demonstrating that evaluating people for the likelihood of FH at the level of PCPs is an effective way to identify patients with this genetic condition and prevent them from developing life‑threatening diseases.
Introduction
Familial hypercholesterolemia (FH) is a group of monogenic diseases with an autosomal mode of inheritance that lead to significantly high low‑density lipoprotein cholesterol (LDL‑C) levels and early‑onset cardiovascular diseases (CVDs). It is the most common monogenic disorder, with an estimated prevalence of 1 in 250 adults in the Polish population.1 Around 140 000 adult patients receiving outpatient care in Poland may have definite or probable FH.2 Among patients hospitalized for acute coronary syndrome, the incidence may be as high as 1 in 23.3 In most cases, FH is associated with a mutation in the LDL receptor (LDLR) gene, and in the remaining cases, with the apolipoprotein B (APOB) gene or gain‑of‑function mutations of the proprotein convertase subtilisin / kexin 9 gene. A mutation causes a lipid disorder with high LDL‑C level from childhood. As a result, this condition leads to early CVD.4 Although the diagnosis of FH does not require genetic testing and can be based on lipid profile, physical examination, and family history, in many countries, including Poland, it is usually diagnosed and managed after a CVD event, such as heart attack or stroke, often at a young age. Most patients are unaware of FH, and do not undertake preventive or therapeutic measures. The lipid profile, including its LDL‑C component, is the primary tool for diagnosing FH. It can be suspected in adults with total cholesterol level at or above 8 mmol/l or LDL‑C level at or above 4.9 mmol/l, who have a family history of premature atherosclerotic disease.4 A practical diagnostic tool is the Dutch Lipid Clinic Network (DLCN) scale.5 A score above 8 points confirms a definitive diagnosis of FH. Scores of 6–8 suggest likely FH, scores of 3–5 indicate possible FH; both need further testing. The final confirmation of FH relies on genetic testing, and the next recommended step is cascade screening of family members. It involves diagnosing the disease in a proband through lipid and molecular testing, followed by lipid profile and DNA analysis of relatives across generations.6
Recently, there has been a growing interest in diagnosing lipid disorders in Poland, as well as in developing programs that increase the frequency and scope of such diagnoses. Currently, lipid profile assessment is part of routine checkups for 6‑year‑old Polish children. In this paper, we present a model of primary care provider (PCP)-initiated screening for FH in the adult Polish population, implemented as part of the Kordian primary prevention program for CVD at the University Hospital in Kraków. We also show the spectrum of FH variants identified in the study group, the characteristics of mutation carriers, and the clinical factors that influence the likelihood of a genetic diagnosis of FH.
Patients and methods
The study was conducted from 2019 to 2023 as part of the project “Prevention of atherosclerosis and heart diseases through education and genetic testing for familial hypercholesterolemia in people with numerous cardiovascular risk factors in the macroregion of south‑eastern Poland (Kordian).” The project was funded by the Polish Ministry of Health under the Operational Program for Knowledge, Education, and Development 2014–2020, cofinanced by the European Social Fund. It aimed to increase awareness and enhance detection rates of CVD among men and women of working age over 18 years, with a particular focus on identifying individuals at a high risk of FH.
The first project stage was conducted by PCPs. During the launch of the Kordian program, an open call was issued to PCPs. To qualify for the project, institutions needed to meet certain criteria, primarily their location in southern Poland (the Lesser Poland, Podkarpackie, or Świętokrzyskie voivodeships). In total, 54 PCPs participated in the program: 9 in provincial capitals, 22 in smaller towns, and 23 in rural areas. The staff of the participating PCPs received training on diagnosing and managing cardiovascular risk factors, including FH, using the DLCN scale, and implementing lipid‑lowering treatments. Patients without prior chronic disease diagnosis who took no medications were eligible, as per the program’s protocol. During the initial visit conducted by the PCP, the patients were examined, and if there were indications to initiate pharmacotherapy for a diagnosed cardiovascular condition, it was started immediately. The study involved 4018 patients without prior CVD, who had interviews, physical examinations, and blood tests, including a complete lipid profile and glucose level measurement. Additionally, the DLCN score was calculated. A total of 1234 individuals with CVD risk factors, such as abnormalities in blood pressure (BP), lipid profile, glucose, or body mass index (BMI), could benefit from the active prevention phase, which included professional counselling in diet, health education, and physical activity, lasting a total of 360 minutes and delivered through an educational voucher.
The individuals who scored at least 6 points on the DLCN scale and those with 3–5 points, with a strongly positive family history of early CVD or high LDL‑C level in children (an additional arbitrary criterion), were also referred to the Department of Cardiology of the University Hospital in Kraków (n = 378). A strongly positive family history was defined as at least 2 episodes of premature CVD in 2 different first‑degree family members or 1 episode before 40 years of age. After ruling out secondary causes of lipid disorders, such as hypothyroidism, poorly controlled diabetes, medications, ketogenic diet, anorexia nervosa, obesity, and kidney or liver diseases, the patients underwent genetic testing for FH. The final qualification and implementation of genetic tests were performed by specialists at the Department of Cardiology. Currently, cascade screening in the families of probands continues as part of our hospital’s routine clinical practice. The FH patients identified via the Kordian program or cascade screening are now receiving regular outpatient medical care. All FH mutation carriers were offered the possibility of further monitoring by the University Hospital. We estimate that current acceptance rate for such care is just over 70% (unpublished data).
The protocol of the Kordian program was reviewed and accepted (8/KBL/OIL/2020) by the Bioethics Committee of the Krakow District Medical Chamber. The study was conducted in accordance with the principles outlined in the Declaration of Helsinki.
Molecular genetic analysis
Genomic DNA was extracted from peripheral blood using the Maxwell RSC kit (Promega GmbH, Madison, Wisconsin, United States), and its quality and quantity were then measured using a Quantus fluorometer (Promega GmbH). Next‑generation sequencing (NGS) analysis of the proband genomic DNA was conducted using the Ampliseq for Illumina on‑demand kit (Illumina, San Diego, California, United States). After preparing the libraries, the samples were amplified and sequenced using the MiSeq sequencer (Illumina). Bioinformatic analyses were conducted using the BaseSpace and Variant Interpreter applications (Illumina) in relation to the human genome hg19. The nomenclature of the identified changes followed the 2016 Human Genome Variation Society. The interpretation of the significance of the detected genetic variants and their connection with the observed disease was classified using current databases, including Varsome, ClinVar, Franklin, GeneBe, expert recommendations, and scientific publications. The presence of pathogenic variants in the probands was confirmed by Sanger sequencing using a 3500 Genetic Analyzer (Thermo Fischer Scientific, Waltham, Massachusetts, United States). The multiplex ligation‑dependent probe amplification (MLPA) test for detecting deletions or duplications in the LDLR gene was carried out using SALSA MLPA Probemix for the LDLR gene (MRC‑Holland, Amsterdam, the Netherlands).
Statistical analysis
The patients with a confirmed pathogenic genetic mutation for FH were compared with those without such confirmation. Categorical variables are shown as numbers and / or percentages. Continuous variables are presented as mean (SD) or median (interquartile range [IQR]). Differences between the groups were analyzed using the t test for normally distributed continuous variables, while the Wilcoxon rank‑sum test was employed for non‑normally distributed continuous variables. The Pearson χ2 test was used to evaluate categorical variables. In the case of ordinal variables with more than 2 values, the distribution of FH pathogenic mutations was compared using the Cochran–Armitage trend test.
Univariable and multivariable logistic regressions identified predictors of the outcome of a genetic test confirming pathogenic FH mutation. The multiple regression model included factors identified by the stepwise regression model with a P value threshold (0.25 to enter, 0.1 to leave). A 2‑sided P value below 0.05 indicated significance. JMP package, version 17.1.0 (JMP Statistical Discovery, Cary, North Carolina, United States) was utilized for all statistical analyses.
Results
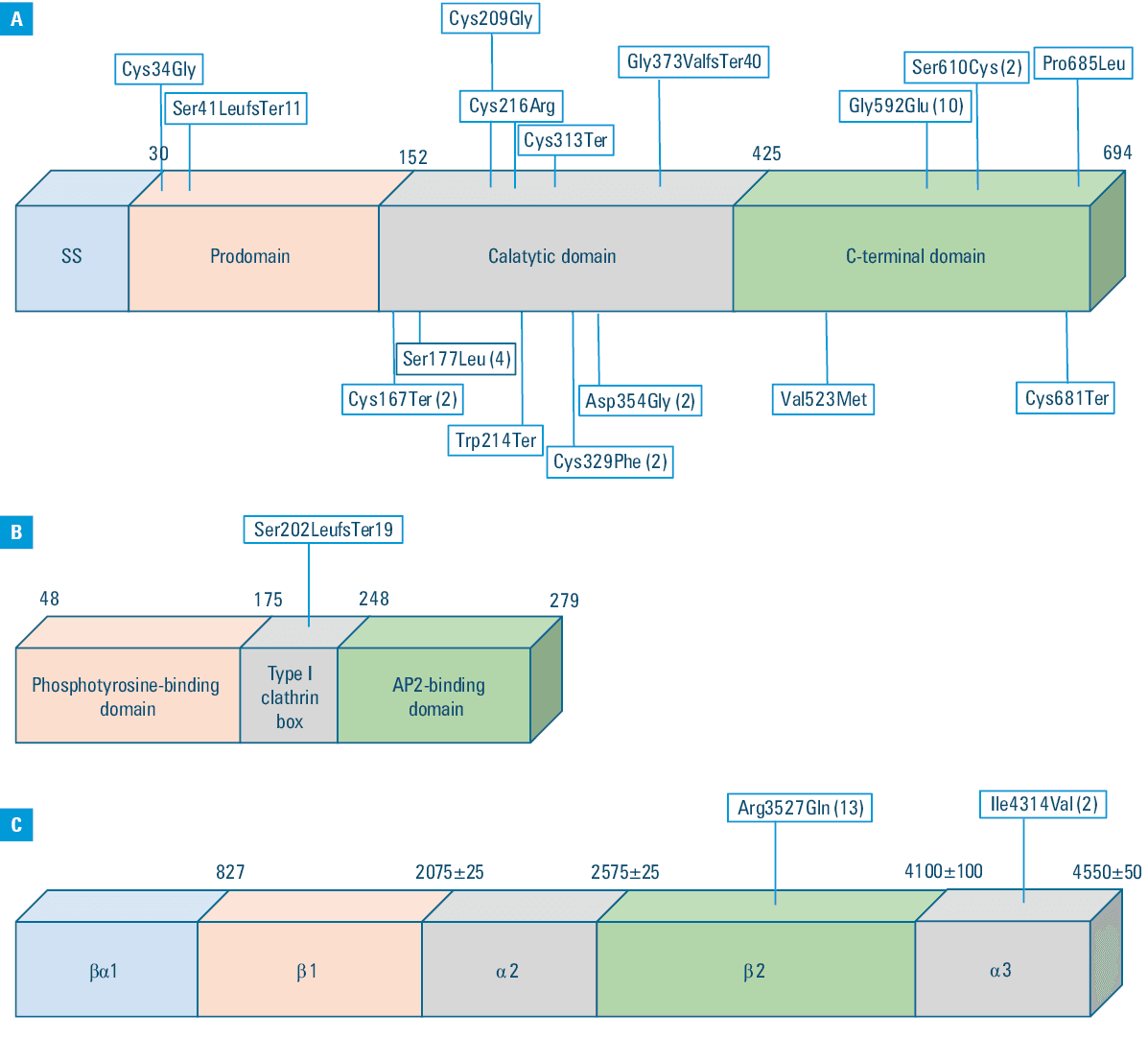
The established criteria for NGS genetic testing were met by 125 patients, representing 33.1% of those referred to the Department of Cardiology. Among them, 51 were men (40.8%). The mean (SD) age of the patients was 46.2 (13.1) years. As many as 97 patients (77.6%) had a DLCN score of 6 or higher at the time of genetic testing qualification. There were 28 patients (22% of the entire group) with a DLCN score of 3–5 points referred for genetic testing. The median (IQR) DLCN score before the genetic test was 6 (6–8) points, and after the test, it increased to 9 (6–14) points. A pathogenic FH mutation was identified in 57 patients (45.6% of the initial cohort), including 6 patients from the DLCN subgroup with 3–5 points (21.4% in this subgroup vs 52.6% among the patients with the score ≥6). Mutations in the LDLR gene were the most commonly identified cause of FH in our patient group. The second most frequently diagnosed mutations were found in the APOB gene. Together, mutations in these 2 genes accounted for over 98% of the pathogenic variants (n = 56). Only 1 mutation was identified in the LDLRAP1 gene (1.8% of the entire cohort). The distribution of the pathogenic mutations within the gene structure is shown in Figure 1A–1C, and detailed characteristics are included in Table 1. All identified mutations have been previously reported by other authors. There were 53 individuals with a pathogenic mutation identified through NGS and 4 by the MLPA test, respectively.
Sample number | Gene | cDNA (HGVS) | Protein (HGVS) | Type of variant | References |
Abbreviations: APOB, apolipoprotein B; HGVS, Human Genome Variation Society; LDLR, low‑density lipoprotein receptor; LDLRAP1, low‑density lipoprotein receptor adaptor protein 1 | |||||
1 | LDLRAP1 | c.603dupC | p.Ser202LeufsTer19 | Exon duplication | Tada et al29 |
2 | LDLR | c.1829C>G | p.Ser610Cys | Amino acid change | Dušková et al30 |
3 | LDLR | rsa[GRCH36] 19p13.2(11078190_11085298) × 1 | Deletion of exons 5–10 | Taylor et al31 | |
4 | LDLR | c.1118delG | p.Gly373ValfsTer40 | Deletion | Rader et al32 |
5 | LDLR | c.1705+1G>A | – | Splice change | Hattori et al33 |
6 | APOB | c.12940A>G | p.Ile4314Val | Amino acid change | Neale et al,34 Radovica‑Spalvina et al35 |
7 | LDLR | c.646T>C | p.Cys216Arg | Amino acid change | Mozas et al36 |
8 | LDLR | c.2054C>T | p.Pro685Leu | Amino acid change | Huijgen et al37 |
9 | LDLR | c.100T>G | p.Cys34Gly | Amino acid change | Meshkov et al38 |
10 | LDLR | c.1829C>G | p.Ser610Cys | Amino acid change | Dušková et al30 |
11 | LDLR | c.501C>A | p.Cys167Ter | Amino acid change (stop codon) | Meshkov et al38 |
12 | LDLR | c.530C>T | p.Ser177Leu | Amino acid change | Banerjee et al39 |
13 | LDLR | c.530C>T | p.Ser177Leu | Amino acid change | Banerjee et al39 |
14 | APOB | c.10580G>A | p.Arg3527Gln | Amino acid change | Fernández‑Higuero et al40 |
15 | APOB | c.10580G>A | p.Arg3527Gln | Amino acid change | Fernández‑Higuero et al40 |
16 | LDLR | c.1775G>A | p.Gly592Glu | Amino acid change | Chmara et al21 |
17 | LDLR | c.530C>T | p.Ser177Leu | Amino acid change | Banerjee et al39 |
18 | APOB | c.10580G>A | p.Arg3527Gln | Amino acid change | Fernández‑Higuero et al40 |
19 | LDLR | c.1567G>A | p.Val523Met | Amino acid change | Hori et al41 |
20 | LDLR | c.501C>A | p.Cys167Ter | Amino acid change (stop codon) | Meshkov et al38 |
21 | LDLR | c.119dupT | p.Ser41LeufsTer11 | Exon duplication | Totoń-Żurańska et al42 |
22 | LDLR | c.641G>A | p.Trp214Ter | Amino acid change | Rader et al32 |
23 | LDLR | c.1061A>G | p.Asp354Gly | Aamino acid change | Chmara et al21 |
24 | APOB | c.10580G>A | p.Arg3527Gln | Amino acid change | Fernández‑Higuero et al40 |
25 | LDLR | c.1775G>A | p.Gly592Glu | Amino acid change | Chmara et al21 |
26 | APOB | c.10580G>A | p.Arg3527Gln | Amino acid change | Fernández‑Higuero et al40 |
27 | LDLR | c.1775G>A | p.Gly592Glu | Amino acid change | Chmara et al21 |
28 | APOB | c.10580G>A | p.Arg3527Gln | Amino acid change | Fernández‑Higuero et al40 |
29 | APOB | c.12940A>G | p.Ile4314Val | Amino acid change | Neale et al,34 Radovica‑Spalvina et al35 |
30 | LDLR | c.1775G>A | p.Gly592Glu | Amino acid change | Chmara et al21 |
31 | LDLR | c.1775G>A | p.Gly592Glu | Amino acid change | Chmara et al21 |
32 | LDLR | c.1775G>A | p.Gly592Glu | Amino acid change | Chmara et al21 |
33 | LDLR | c.939C>A | p.Cys313Ter | Amino acid change | Chmara et al21 |
34 | LDLR | rsa[GRCH36] 19p13.2(11078200_11085310) × 1 | Deletion of exons 5–10 | Taylor et al31 | |
35 | LDLR | c.625T>G | p.Cys209Gly | Amino acid change | Meshkov et al38 |
36 | APOB | c.10580G>A | p.Arg3527Gln | Amino acid change | Fernández‑Higuero et al40 |
37 | LDLR | c.986G>T | p.Cys329Phe | Amino acid change | Meshkov et al38 |
38 | APOB | c.10580G>A | p.Arg3527Gln | Amino acid change | Fernández‑Higuero et al40 |
39 | APOB | c.2981C>T | p.Pro994Leu | Amino acid change | Alves et al43 |
40 | LDLR | c.1775G>A | p.Gly592Glu | Amino acid change | Chmara et al21 |
41 | APOB | c.10580G>A | p.Arg3527Gln | Amino acid change | Fernández‑Higuero et al40 |
42 | LDLR | c.941–2A>G | – | Change in intron | Han et al44 |
43 | APOB | c.10580G>A | p.Arg3527Gln | Amino acid change | Fernández‑Higuero et al40 |
44 | LDLR | c.1775G>A | p.Gly592Glu | Amino acid change | Chmara et al21 |
45 | APOB | c.10580G>A | p.Arg3527Gln | Amino acid change | Fernández‑Higuero et al40 |
46 | LDLR | c.1705+1G>A | – | Splice change | Hattori H et al33 |
47 | LDLR | rsa[GRCH36] 19p13.2(11074272_11079083) × 3 | Duplication of exons 3–6 | Chmara et al21 | |
48 | LDLR | c.1061A>G | p.Asp354Gly | Amino acid change | Chmara et al21 |
49 | LDLR | c.2043C>A | p.Cys681Ter | Amino acid change | Sharifi et al,19
Rader et al32 |
50 | LDLR | c.1775G>A | p.Gly592Glu | Amino acid change | Chmara et al21 |
51 | APOB | c.10580G>A | p.Arg3527Gln | Amino acid change | Fernández‑Higuero et al40 |
52 | LDLR | c.530C>T | p.Ser177Leu | Amino acid change | Banerjee et al39 |
53 | LDLR | rsa[GRCH36] 19p13.2(11074272_11079083) × 3 | Duplication of exons 3–6 | Chmara et al21 | |
54 | LDLR | c.986G>T | p.Cys329Phe | Amino acid change | Meshkov et al38 |
55 | LDLR | c.1775G>A | p.Gly592Glu | Amino acid change | Chmara et al21 |
56 | APOB | c.10580G>A | p.Arg3527Gln | Amino acid change | Fernández‑Higuero et al40 |
57 | LDLR | c.941–13T>A | – | Change in intron | Chmara et al21 |
We compared the patients with a confirmed pathogenic FH mutation and those without such a mutation. The patients with genetically confirmed FH were younger (41 vs 49 y), had lower BMI (24.59 vs 27.22 kg/m2), and lower diastolic BP (DBP), as well as numerically lower systolic BP. The patients with a confirmed mutation used statins similarly often but took ezetimibe more frequently, while utilization of β-blockers and angiotensin‑converting enzyme inhibitors (ACEIs)/sartans was lower than in the comparator group. Table 2 presents a comparison of essential demographic and clinical data between the study groups.
Parameter | FH not confirmed (n = 68) | FH confirmed (n = 57) | P value | |
Data are presented as number (percentage) or median (interquartile range) unless indicated otherwise.
a Pearson χ2 test; b Wilcoxon test; c t test
Abbreviations: ACEI, angiotensin‑converting enzyme inhibitor; AF, atrial fibrillation; BMI, body mass index; DBP, diastolic blood pressure; DM, diabetes mellitus; FH, familial hypercholesterolemia; HR, heart rate; SBP, systolic blood pressure | ||||
Sex | Men | 31 (45.6) | 20 (35.1) | 0.23a |
Women | 37 (54.4) | 37 (64.9) | ||
Age, y | 49 (39.25–61.75) | 41 (34–52) | 0.002b | |
Weight, kg, mean (SD) | 80.36 (10.65) | 70.33 (12.96) | 0.004c | |
Height, cm, mean (SD) | 171.82 (7.32) | 168.83 (8.99) | 0.19c | |
BMI, kg/m2, mean (SD) | 27.22 (3.31) | 24.59 (3.61) | 0.009c | |
SBP, mm Hg, mean (SD) | 136.25 (17.36) | 131.07 (21.67) | 0.15c | |
DBP, mm Hg, mean (SD) | 88.06 (10.04) | 83.75 (12.78) | 0.04c | |
HR, bpm, mean (SD) | 73.78 (11.79) | 76.68 (12.64) | 0.2c | |
Smoking | 17 (25) | 12 (21.1) | 0.6a | |
AF | 1 (1.5) | 1 (1.8) | 0.89a | |
DM | 3 (4.4) | 2 (3.5) | 0.8a | |
Hypertension | 25 (36.8) | 12 (21.1) | 0.06a | |
Peptic ulcer disease | 4 (5.9) | 0 | 0.06a | |
Thyroid disease | 9 (13.2) | 6 (10.5) | 0.64a | |
Kidney disease | 0 | 1 (1.8) | 0.27a | |
Medications | Statins | 58 (86.6) | 48 (84.2) | 0.71a |
Ezetimibe | 48 (71.6) | 48 (84.2) | 0.1a | |
ACEI/sartans | 20 (29.9) | 8 (14) | 0.04a | |
β-Blockers | 15 (22.4) | 3 (5.3) | 0.007a | |
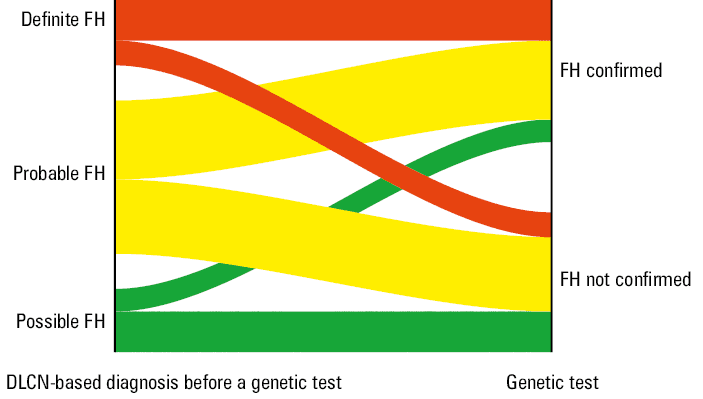
Analyzing the DLCN classification scores obtained by the patients in both study groups before the genetic test, we observed higher scores in the group in which the genetic test confirmed the pathological FH mutation. However, both groups scored a very similar number of points on the DLCN scale for the LDL‑C level. Conversely, we found several differences in the patient clinical features. First, in the group in which FH was confirmed, more than twice as many patients had first‑degree relatives with LDL‑C levels exceeding 4.9 mmol/l. Moreover, over 3 times as many patients with genetically confirmed FH exhibited tendon xanthomas. Among the individuals with a confirmed genetic basis for FH, more patients had xanthomas of the eyelids and corneal limbus, but this difference was not significant. Detailed data on DLCN point distribution are provided in Table 3. The distribution of patients into study groups based on the initial DLCN assessment results is illustrated in Figure 2. We also observed some differences in biochemical parameters between the study groups, as presented in Table 4.
Parameter | FH not confirmed (n = 68) | FH confirmed (n = 57) | P value | |
Data are presented as number (percentage) or median (interquartile range).
a Wilcoxon test; b Cochran–Armitage trend test; c Pearson χ2 test
Abbreviations: AC, arcus cornealis; CAD, coronary artery disease; DLCN, Dutch Lipid Clinic Network; LDL‑C, low‑density lipoprotein cholesterol; PAD, peripheral artery disease; TX, tendon xanthomas; others, see Table 2 | ||||
DLCN score for clinical data, points | 1 (0–1) | 1 (1–2) | 0.001a | |
0 | 23 (33.8) | 8 (14) | 0.006b | |
1 | 39 (57.4) | 34 (59.7) | ||
2 | 2 (2.9) | 5 (8.8) | ||
5 | 0 | 1 (1.8) | ||
6 | 2 (2.9) | 0 | ||
7 | 2 (2.9) | 7 (12.3) | ||
8 | 0 | 2 (3.5) | ||
First‑degree relative with premature CAD/PAD | 33 (48.5) | 23 (40.4) | 0.36c | |
First‑degree relative with LDL‑C >4.9 mmol/l | 19 (27.9) | 41 (71.9) | <0.001c | |
First‑degree relative with TX or AC | 0 | 2 (3.5) | 0.12c | |
Child with LDL‑C >3.9 mmol/l | 2 (2.9) | 5 (8.8) | 0.16c | |
TX | 3 (4.4) | 9 (15.8) | 0.03c | |
Xanthomas of the eyelids | 2 (2.9) | 4 (7) | 0.29c | |
AC <45 y | 0 | 2 (3.5) | 0.12c | |
DLCN points for LDL‑C | 5 (3–8) | 5 (5–8) | 0.11a | |
3 | 18 (26.5) | 4 (7) | 0.15b | |
5 | 30 (44.1) | 35 (61.4) | ||
8 | 20 (29.4) | 18 (31.6) | ||
DLCN score before a genetic test, points | Median | 6 (5–8) | 6 (6–9) | 0.01a |
Possible FH | 22 (32.4) | 6 (10.5) | 0.005b | |
Probable FH | 34 (50) | 34 (59.7) | ||
Definite FH | 12 (17.7) | 17 (29.8) | ||
DLCN score after a genetic test, points | Median | 6 (5–8) | 14 (14–17) | <0.001a |
Possible FH | 22 (32.4) | 0 | <0.001b | |
Probable FH | 34 (50) | 0 | ||
Definite FH | 12 (17.7) | 57 (100) | ||
Parameter | FH not confirmed (n = 68) | FH confirmed (n = 57) | P value |
a Wilcoxon test; b Pearson χ2 test; c Cochran–Armitage trend test
SI conversion factors: to convert ALT and AST to µkat/l, multiply by 0.0167
Abbreviations: ALT, alanine transaminase; AST, aspartate transaminase; CK, creatine kinase; CRP, C‑reactive protein; eGFR, estimated glomerular filtration rate; H, above normal range; HDL‑C, high‑density lipoprotein; L, below normal range; N, within normal range; TC, total cholesterol; TG, triglyceride; TSH, thyroid‑stimulating hormone; others, see Tables 2 and 3 | |||
TC, mmol/l, median (IQR) | 9.04 (8.3–10.62) | 10.04 (9.16–11.2) | 0.01a |
LDL‑C, mmol/l, median (IQR) | 7.01 (6.31–8.6) | 7.77 (7.01–8.72) | 0.02a |
TG, mmol/l, median (IQR) | 1.6 (1.21–2.22) | 1.3 (1.02–1.71) | 0.03a |
HDL‑C, mmol/l, median (IQR) | 1.4 (1.27–1.83) | 1.54 (1.21–1.82) | 0.53a |
Lipoprotein(a), g/l | |||
Median (IQR) | 0.24 (0.09–0.83) | 0.18 (0.1–0.51) | 0.68a |
H/N, n (%) | 29 (43.3)/38 (56.7) | 23 (40.4)/34 (59.7) | 0.74b |
TSH, μIU/ml | |||
Median (IQR) | 1.62 (1.21–2.37) | 1.62 (1.13–1.93) | 0.47a |
H/N/L, n (%) | 0/65 (97)/2 (3) | 1 (1.8)/56 (98.3)/0 | 0.89c |
AST, U/I | |||
Median (IQR) | 29 (24–36.5) | 26 (23–32) | 0.08a |
H/N, n (%) | 3 (4.6)/63 (95.5) | 3 (5.3)/54 (94.7) | 0.85b |
ALT, U/I | |||
Median (IQR) | 33 (22–53) | 24 (18–38) | 0.008a |
H/N/L, n (%) | 18 (26.9)/ 48 (71.6)/1 (1.5) | 6 (10.5)/ 48 (84.2)/3 (5.3) | 0.04c |
CK, U/l | |||
Median (IQR) | 114 (78.25–197.75) | 107.5 (81.25–160.75) | 0.57a |
H/N, n (%) | 5 (7.6)/61 (92.4) | 5 (8.9)/51 (91.1) | 0.79b |
Creatinine, mmol/l | |||
Median (IQR) | 69.7 (59.5–78.3) | 67.2 (58.9–77.35) | 0.41a |
H/N/L, n (%) | 1 (1.5)/ 46 (68.7)/20 (29.9) | 1 (1.8)/ 33 (58.9)/22 (39.3) | 0.53c |
eGFR, ml/min/1.73 m2 | |||
Median (IQR) | 90 (87–90) | 90 (90–90) | 0.25a |
N/L, n (%) | 67 (100)/0 | 55 (98.2)/1 (1.8) | 0.27b |
Glucose, mmol/l | |||
Median (IQR) | 5.15 (4.8–5.54) | 4.87 (4.73–5.17) | 0.01a |
H/N, n (%) | 11 (16.4)/56 (83.6) | 4 (7)/53 (93) | 0.11b |
CRP, mg/l | |||
Median (IQR) | 1.28 (1–2.23) | 1 (1–1.8) | 0.08a |
H/N, n (%) | 6 (9)/61 (91) | 3 (5.3)/54 (94.7) | 0.43b |
The univariable analysis identified 5 factors linked to pathogenic FH mutations. The higher were the values of 3 of these factors—age, BMI, and DBP—the lower the likelihood of confirming a pathogenic mutation. Conversely, the total score in the DLCN classification, relating to the patient clinical characteristics but excluding points for the LDL‑C level and considering having a first‑degree relative with an LDL‑C level above 4.9 mmol/l, showed a positive relationship with the mutation frequency. In the multivariable analysis, 2 factors remained significant: BMI, where higher levels reduced the mutation risk, and having a relative with high LDL‑C level, increasing the risk over 11‑fold. Detailed data are provided in Table 5.
Parameter | Univariable, OR (95% CI) | P value | Multivariable, OR (95% CI) | P value |
Age, per 1 y | 0.96 (0.93–0.98) | 0.001 | – | – |
BMI, per 1 kg/m2 | 0.8 (0.66–0.96) | 0.02 | 0.76 (0.61–0.95) | 0.006 |
First‑degree relative with LDL‑C >4.9 mmol/l | 6.61 (3.02–14.47) | <0.001 | 11.37 (2.69–48.12) | <0.001 |
DBP, per 1 mm Hg | 0.97 (0.94–0.99) | 0.04 | – | – |
DLCN for clinical data | 1.32 (1.06–1.62) | 0.005 | – | – |
Discussion
Genetic testing for FH in Poland, similarly to many other countries, is mainly performed as part of a secondary cardiovascular prevention after a young person has experienced heart attack or stroke, particularly when a high LDL‑C level has been detected. It is also sometimes initiated as a cascade screening within the proband’s family. In this paper, we described a patient pathway model initiated by the PCP that successfully led to the genetic diagnosis of FH in a large proportion of referred patients with no prior history of CVD, thereby enabling pharmacologic treatment before any event occurred. The key element of this model is the application of the DLCN scale by the PCP, followed by referral to a specialist center, with the final decision on genetic testing made by specialists with access to NGS‑based genetic testing. Below, we discuss the details of the proposed model, the range of identified FH mutations, and the clinical features of the mutation carriers.
Model performance
Within the primary cardiovascular prevention Kordian program, a high FH risk group was identified and referred to a tertiary center for comprehensive diagnostics, including genetic testing. The patients with confirmed pathogenic mutations responsible for FH accounted for 4.6% of the entire population included in the Kordian program, 15.1% of the population referred to the reference coordinating center, and 45.6% of the population selected by specialists for genetic testing. This shows reasonable specificity of the proposed algorithm. Given the estimated prevalence of FH in the Polish population of 0.4%, it can be concluded that the Kordian program initially identified a group of patients with nearly a 12‑fold higher risk of FH than the general population. Furthermore, PCP narrowed down the population to individuals with the risk increased by more than 38 times. The specialists referring the participants for genetic tests helped identify a population with a 114 times higher FH risk than the general Polish population. Projects similar to Kordian, involving PCP and centers for lipid disorder diagnosis and treatment, were conducted in other European countries, with detection rates of pathogenic FH mutations similar to ours. For example, a program with the acronym ARIAN was implemented in Spain. After excluding secondary causes of high LDL‑C levels (above 6.5 mmol/l), patients with DLCN score of at least 6 points were referred for genetic testing. Under the ARIAN program, 153 patients were referred for genetic testing; 67 were confirmed to have a pathogenic FH mutation.7 The patients with confirmed FH accounted for nearly 44% of those tested, allowing the program to identify high‑risk FH individuals as effectively as Kordian. Additionally, researchers from the United Kingdom found pathogenic FH mutations in 36.5% of 635 patients who met the Simon Broome FH criteria (definite and probable FH).8 Among patients in the Korean population eligible for genetic testing under the same classification, 32% were found to have pathogenic FH mutations in a cohort of 97 individuals.9 In a London hospital, among 204 patients qualifying for genetic testing under the Simon Broome criteria, nearly 55% had pathogenic FH mutations.10 In our group, this value of 45.6% corresponds with that reported by researchers in London. This Figure ranks among the highest recorded in the literature, supporting the assumptions behind the methodology used to select patients for genetic testing in the Kordian program.
Data from the Polish population suggest that over half of the patients with a probable diagnosis of FH have been diagnosed and treated solely as part of secondary prevention.2 Therefore, developing algorithms for primary prevention, such as those implemented in the Kordian program, is essential for FH mutation carriers. We demonstrated that an integrated primary care–specialist pathway using clinical DLCN scoring effectively identifies FH patients before cardiovascular events occur. Notably, the PCPs conducted DLCN score assessments in a large cohort of patients. The predicted cost of performing a DLCN assessment in the PCP outpatient setting is not high, as it relies on medical history, physical examination, and lipid level measurements; genetic testing is not essential for diagnosing FH. PCPs should have access to a reference center where FH genetic testing can be performed under the National Health Fund contract, and the number of such centers is increasing.
Genetic findings
The first gene linked to FH was the LDLR gene, discovered in the 1970s.11 In our group, 40 patients (70%) had mutations in this gene, aligning with data from other populations. Genetic screening in the Dutch population found that about 67% of FH patients carry an LDLR mutation.12 In the French population, this percentage was 73.9%,13 in the Taiwanese population 89.89%,14 and it exceeded 94% in an Italian cohort.15
The second gene linked to FH development is APOB.16 In our group, 28% of the patients had APOB mutations, a rate higher than in other cohorts. In studies on the Dutch population, this percentage was approximately 8%.17 A slightly higher mutation rate in this gene was reported in Asian populations, occurring in up to 20% of individuals with a confirmed genetic FH background.18 This discrepancy may be due to our study analyzing the entire APOB gene, rather than the exon 26 fragment containing the p.Arg3527Gln mutation.19 Our observations align with data on FH mutations in Poland, where LDLR mutations are the most common.19-21
Clinical features
The patients with a confirmed pathogenic FH mutation had slightly different clinical characteristics than those without such a confirmation. The patients with a confirmed genetic diagnosis of FH were younger and had lower BMI than those whose diagnosis of FH was not confirmed. The latter showed coexistence of other metabolic phenotypes, as indicated by higher values of triglycerides, alanine transaminase, glucose, and BP. Similar observations were published in 2024 in Ireland, where patients with lipid disorders confirmed to have the genetic basis of FH were less likely to be diagnosed with hypertension, but more likely to have a positive family history of premature CVD.22 Data from the New Zealand population also indicated that patients with high LDL‑C level and genetic FH diagnosis were, similarly to our population, younger and more likely to have relatives with high LDL‑C levels.23 PCPs started lipid‑lowering therapy with a similar frequency in both our study subgroups. Conversely, ACEIs/sartans were prescribed twice as often in the patients without FH mutations, while β-blockers were used nearly 4 times more frequently as compared with the genetically confirmed FH group. This may indirectly imply that the patients without confirmed FH had more comorbidities, such as arterial hypertension and overweight or obesity. This suggests that their high LDL‑C level is rather of a polygenic than monogenic origin and is part of broader metabolic syndrome.
Analysis of the features that significantly influenced the likelihood of confirming the genetic diagnosis of FH showed that, in this study group, a very important factor was having a first‑degree relative with an LDL‑C level exceeding 4.9 mmol/l. This observation aligns with data from some other available publications.24,25 A very high LDL‑C level in a relative is a factor often recognized in the literature as significantly increasing the risk of FH. Interestingly, this factor serves as an alternative element to the DLCN risk assessment scales for FH. One such scale was established by the American Heart Association (AHA) and is used to assess the likelihood of FH in specific patients.25 Furthermore, according to this AHA definition, a patient in whom this factor has been identified and who is concurrently diagnosed with an elevated LDL‑C level, also above 4.9 mmol/l, can be diagnosed with FH. Increased LDL‑C level in a relative is also 1 of the 5 criteria of the Simon Broome Familial Hypercholesterolemia Register diagnostic criteria for FH.26 This classification does not account for LDL‑C, but rather for total cholesterol; the limit for FH diagnosis was set at 7.5 mmol/l. Importantly, this classification also includes a criterion for the occurrence of CVD in slightly younger patients than in the case of the DLCN scale; the criterion is met by second‑line relatives who were diagnosed with heart attack under the age of 50 years. Specialists qualifying the patients for genetic testing during the Kordian program adopted a similar reduction in age as a factor significantly increasing the risk of FH in a patient, however, in this case, the age was reduced to 40 years, resulting in the frequency of genetic confirmation of the diagnosis of FH approaching 50% of the population referred for genetic testing, even if this group included patients with 3 points on the DLCN scale. This result emphasizes the crucial importance of family history, as well as the clinical expertise of the specialists working within the Kordian program.
FH is an autosomal‑dominant trait; therefore, an equal prevalence of mutations in both sexes is expected. Interestingly, there were more women than men in the final Kordian cohort; we believe this finding reflects selection bias and likely greater willingness among women to participate in genetics studies.26
Limitations
This study has several shortcomings that warrant discussion. First, the FH screening was conducted using the Kordian program’s primary‑prevention assumptions; therefore, patients with a prior cardiovascular event were excluded. Additionally, the percentage of individuals from the initial Kordian population with identified FH mutations was unexpectedly high (4.6%). Selection bias is the most plausible explanation for this result. It seems that the PCPs were more inclined to encourage patients with particularly high LDL‑C levels and a positive history of CVD to participate. Simultaneously, these patients were probably more willing to accept such an invitation. It may also be worth emphasizing that this is not an epidemiologic study, and that some degree of bias is likely unavoidable. The study lacks information on patient rates of decline to participate, which could provide additional insight into the magnitude of potential selection bias. Furthermore, an inclusion criterion of having 3–5 points on the DLCN scale, along with a strongly positive family history of early CVD or high LDL‑C level in children, was an arbitrary decision based on the clinical judgement of the study team and was not evidence‑based. Additionally, our study did not confirm the pathogenic nature of the identified mutations through functional experiments or by examining their segregation with the FH phenotype within families. Nonetheless, the fact that all of them were classified as pathogenic in the genetic databases and previously reported as FH‑causing variants strongly supports their causal role. Furthermore, the program did not gather detailed data on all initial study participants, including DLCN classification points.
Conclusions
Collaboration between PCPs and specialists with access to genetic testing, guided by a well‑structured algorithm using the DLCN scale, may enable effective identification of patients with FH mutations before the onset of CVD. The results of this study could be valuable for future planning of a nationwide program to screen for FH in adults and support the implementation of primary cardiovascular prevention in this very high‑risk group of mutation carriers. We suggest that PCPs across Poland widely adopt the DLCN scale in their practice, including individuals before a CVD diagnosis, as it is supported by the presented data and aligns with current international diagnostic guidelines.27 This, together with the recent introduction of compulsory screening for high LDL‑C level in 6‑year‑old children in Poland,28 should accelerate the identification of FH mutation carriers in our society.
- Pająk A, Szafraniec K, Polak M, et al. Prevalence of familial hypercholesterolemia: a meta‑analysis of six large, observational, population‑based studies in Poland. Arch Med Sci. 2016; 12: 687‑696. | Crossref
- Chlebus K, Cybulska B, Gruchała M, et al. Prevalence, diagnosis, and treatment of familial hypercholesterolaemia in outpatient practices in Poland. Kardiol Pol. 2018; 76: 960‑967. | Crossref
- Bobrowska B, Zasada W, Rajtar‑Salwa R, et al. Prevalence of familial hypercholesterolemia in patients with acute coronary syndromes, Kardiol Pol. 2019; 77: 475‑477. | Crossref
- Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013; 34: 3478‑3490a. | Crossref
- Defesche JC, Lansberg PJ, Umans‑Eckenhausen MA, Kastelein JJ. Advanced method for the identification of patients with inherited hypercholesterolemia. Semin Vasc Med. 2004; 4: 59‑65. | Crossref
ARTICLE INFORMATION