A 50‑year‑old man presented with a 10‑day history of abdominal discomfort. He underwent splenectomy at the age of 9 years for unexplained massive splenomegaly; preoperative platelet counts were unavailable. His medical history included right femoral head avascular necrosis diagnosed 20 years earlier and 3 hospitalizations for bleeding duodenal ulcers. No easy bruising or growth retardation was reported in childhood. Family history was unremarkable.
Physical examination showed a firm, nontender liver, palpable 8 cm below the right costal margin. Laboratory test results indicated thrombocytosis (platelets, 449 × 109/l; reference range [RR], 100–300 × 109/l), elevated liver enzymes, hypoalbuminemia (albumin, 34 g/l; RR, 35–55 g/l), and a markedly elevated serum ferritin concentration (5676 ng/ml; RR, 30–400 ng/ml). Serum protein electrophoresis, immunofixation, and urine Bence–Jones protein tests were negative, excluding multiple myeloma. Abdominal magnetic resonance imaging (MRI) showed diffuse hepatomegaly with heterogeneous T2 hyperintensity, attenuated intrahepatic vessels, and an empty splenic fossa (Figure 1A). Diffusion‑weighted imaging identified multiple patchy hyperintense lesions in the liver (Figure 1B). Postcontrast images demonstrated marked heterogeneous enhancement, with some areas remaining nonenhancing throughout all phases (Figure 1C–1E). Computed tomography showed avascular necrosis of the right femoral head and multiple vertebral endplate depressions (Figure 1F). Bone marrow biopsy indicated typical Gaucher cells with “wrinkled paper” cytoplasm. The diagnosis was confirmed by considerably reduced leukocyte β-glucosidase activity (2.74 nmol/l/h; RR >6.8 nmol/l/h). Genetic testing identified a heterozygous pathogenic variant c.1448T>C (p.Leu483Pro) in the glucocerebrosidase (GBA) gene. Enzyme replacement therapy with imiglucerase (60 U/kg every 2 weeks) was initiated. After 3 months, the patient reported improved energy and decreased liver enzymes and ferritin.
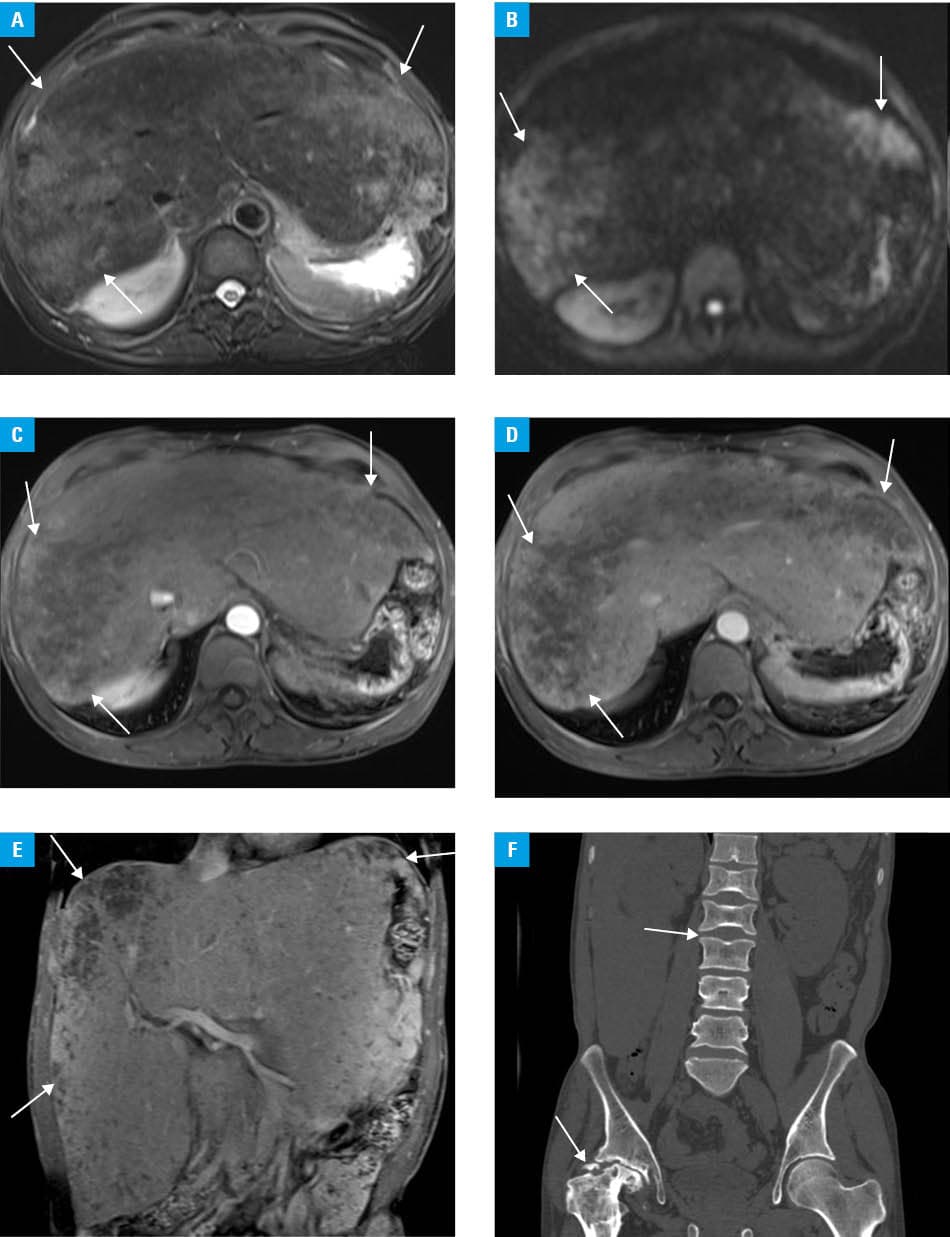
Although hepatosplenomegaly, cytopenia, and bone disease constitute the classic triad, diagnosis is often delayed for years or even decades.1,2 This case illustrates a 41‑year diagnostic delay in type 1 Gaucher disease. Childhood splenectomy removed a key diagnostic clue and led to postoperative thrombocytosis, further confounding the presentation. Progressive hepatomegaly and skeletal complications—femoral avascular necrosis and vertebral depressions—created a misleading clinical picture. Although bone marrow Gaucher‑like cells are suggestive, differentiation from chronic myeloid leukemia and multiple myeloma is required. Multiple myeloma was excluded due to negative urine and serum protein workup. Vertebral endplate depressions on MRI were consistent with skeletal involvement. Markedly elevated ferritin levels provided an additional biochemical indication. Heterogeneous hepatic density warrants differentiation from Gaucheroma and hepatocellular carcinoma, requiring regular imaging surveillance. Genetic testing showed a heterozygous pathogenic GBA variant (c.1448T>C, p.Leu483Pro), a known mutation that reduces β-glucosidase activity. The patient’s mild phenotype was consistent with carrying of at least 1 mild mutation. Although p.Leu483Pro variant is pathogenic, clinical manifestations may vary depending on the second allele. Due to methodological limitations, large deletions or duplications—if present—would not have been detected. This mutation is among the most common GBA variants in the Chinese population.3
In adults with unexplained hepatomegaly, childhood splenectomy, and skeletal complications (avascular necrosis or vertebral collapse), type 1 Gaucher disease should be considered. Splenectomy may obscure the underlying disease and delay diagnosis. A low threshold for bone marrow biopsy and β-glucosidase activity testing is recommended to enable early diagnosis and timely enzyme replacement therapy.
- Linari S, Castaman G. Hemostatic abnormalities in Gaucher disease: mechanisms and clinical implications. J Clin Med. 2022; 11: 6920. | Crossref
- Samonenko N, Olkhovych N, Okhotnikova O, et al. Liver involvement in Gaucher disease type I: a retrospective single‑center study from Ukraine. Front Med (Lausanne). 2025; 12: 1610755. | Crossref
- Subspecialty Group of Endocrinologic, Hereditary and Metabolic Diseases, the Society of Pediatrics, Chinese Medical Association; Subspecialty Group of Hematology, the Society of Pediatrics, Chinese Medical Association; Society of Medical Genetics, Chinese Medical Association; China Alliance for Rare Diseases. Expert consensus on diagnosis and treatment of pediatric Gaucher disease (2021) [in Chinese]. Zhonghua Er Ke Za Zhi. 2021; 59: 1025‑1031. | Crossref
ARTICLE INFORMATION