Biologic and targeted synthetic treatment of SAPHO syndrome: systematic literature review
Key words: biologic disease-modifying antirheumatic drugs, SAPHO syndrome, systematic review, targeted synthetic disease-modifying antirheumatic drugs, treatment



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Biologic and targeted synthetic treatment of SAPHO syndrome: systematic literature review
Introduction: SAPHO syndrome is a rare autoimmune / autoinflammatory disorder with no approved therapies or standardized management guidelines. Biologic and targeted synthetic disease‑modifying antirheumatic drugs (b/tsDMARDs) are increasingly used off‑label.
Objectives: This review aimed to systematically evaluate the evidence for the efficacy of b/tsDMARDs in SAPHO.
Patients and methods: A PRISMA‑guided search of PubMed database was conducted through March 31, 2025. Studies of any design reporting outcomes of b/tsDMARDs in patients with SAPHO fulfilling the modified Kahn criteria were included. Exclusion criteria were non‑English language, inaccessible full text, and insufficient treatment details. Data on patient characteristics, drug class, follow‑up duration, and treatment response were extracted. As all the included studies were case reports or case series, the risk of bias was not assessed. Treatment responses were summarized using descriptive statistics.
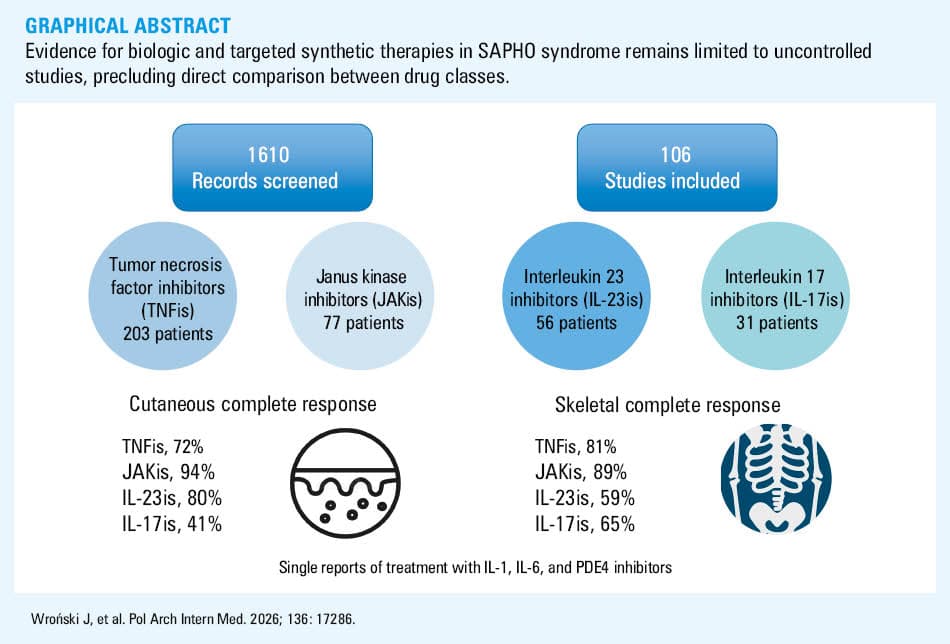
Results: Of 1610 screened records, 106 studies comprising 385 instances of b/tsDMARD use met the inclusion criteria; no randomized controlled trials were identified. Tumor necrosis factor inhibitors (TNFis) were used in 203 patients, with complete skeletal and skin responses in 81% and 72%, respectively. The use of Janus kinase inhibitors (JAKis) in 77 patients was associated with complete response rates of 89% (skeletal) and 94% (skin). Other b/tsDMARD classes—including interleukin 17 (IL‑17), IL‑23, IL‑1, and IL‑6 inhibitors, as well as phosphodiesterase‑4 inhibitors—demonstrated variable and generally lower efficacy.
Conclusions: Evidence for b/tsDMARD efficacy in SAPHO remains limited to low‑quality, uncontrolled studies with substantial heterogeneity and probable publication bias. TNFis and JAKis are among the most consistently effective therapies reported in the available literature, but high‑quality research, including randomized trials or prospective registries, is urgently needed to inform evidence‑based management.
What's new?
This review brings together the largest body of clinical experience to date on modern treatments for SAPHO syndrome, a rare and often difficult‑to‑manage condition lacking formal guidelines. By analyzing outcomes from over 100 published reports, we show that some therapies commonly used in other rheumatic diseases—particularly tumor necrosis factor inhibitors and Janus kinase inhibitors—may provide meaningful relief of skeletal and skin symptoms when other treatments fail. Other targeted drugs, such as interleukin 17 or interleukin 23 inhibitors, show more variable benefit, highlighting the need to match therapy to individual patient profiles. This paper underscores the urgent need for coordinated research efforts, including prospective registries and randomized clinical trials that can transform fragmented case‑based knowledge into evidence‑based care.
Introduction
SAPHO syndrome (synovitis, acne, pustulosis, hyperostosis, and osteitis) is a rare disease (estimated incidence in white patients about 1:10 000; ORPHA code, 793) characterized by the coexistence of osteoarticular inflammation and neutrophilic skin disease, most commonly palmoplantar pustulosis and acne.1 The etiology remains unclear, but infectious triggers, genetic predisposition, and dysregulated innate and adaptive immune responses appear to contribute to the disease development.2 Due to frequent axial involvement and associations with psoriasis and inflammatory bowel disease, SAPHO syndrome is conventionally classified as an autoimmune disease within the spondyloarthritis (SpA) spectrum. On the other hand, neutrophil hyperactivity, reduced level of natural killer cells, elevated levels of proinflammatory cytokines, and clinical similarities with several monogenic autoinflammatory syndromes may indicate autoinflammatory origin.3 Typical manifestations include anterior chest wall osteitis—particularly of the sternoclavicular joints—sacroiliitis, spinal inflammation, palmoplantar pustulosis, peripheral arthritis, and severe acne.4-8 The most widely used classification system, that is, the 2003 modified Kahn criteria,1 also encompass chronic recurrent multifocal osteomyelitis (CRMO), the pediatric form of chronic nonbacterial osteomyelitis, as part of the SAPHO spectrum.
Recently, international expert consensus recommendations9 have proposed the term adult chronic nonbacterial osteomyelitis (CNO) to harmonize terminology for inflammatory bone disease in adults and align it with the pediatric CNO/CRMO spectrum. However, much of the existing clinical literature—including most therapeutic reports—has historically used the broader concept of SAPHO syndrome, which encompasses both osteoarticular inflammation and associated neutrophilic skin disease. As the studies included in this review predominantly employ the SAPHO terminology and frequently describe patients with combined osteoarticular and cutaneous manifestations, we retain the term SAPHO in the present analysis while acknowledging its conceptual overlap with adult CNO.
SAPHO syndrome substantially impairs quality of life through chronic pain and functional limitations, and may lead to local bone destruction, most often in the sternoclavicular region.8 Despite this burden, no approved therapies or standardized care guidelines exist.10,11 Due to the rarity of the condition, treatment recommendations are based almost entirely on case reports and single‑center experiences. The only randomized clinical trial (RCT) to date evaluated pamidronate vs placebo in patients with CNO, but probably due to the small sample size, not all end points reached significance.12
Conventional treatments used in other conditions from the SpA spectrum—nonsteroidal anti‑inflammatory drugs (NSAIDs), conventional synthetic disease‑modifying antirheumatic drugs (DMARDs; eg, methotrexate, sulfasalazine), and systemic or local glucocorticoids—are commonly used in SAPHO,4-6,8 yet only about one‑third of the patients achieve complete remission.13 These limitations have prompted growing interest in biologic and targeted synthetic DMARDs (b/tsDMARDs).
This therapeutic rationale is supported by accumulating evidence of dysregulated innate and adaptive immune signaling in SAPHO pathogenesis. Studies of serum cytokine profiles, lesional tissue, and experimental models suggest activation of neutrophil‑driven inflammation together with aberrant osteo‑immune interactions involving tumor necrosis factor (TNF), interleukin 1β (IL‑1β), IL‑6, and the IL‑23/IL‑17 axis.2,3,10,11 TNF promotes osteitis and synovitis through recruitment of neutrophils, activation of macrophages and T cells, and stimulation of osteoclast differentiation. IL‑1β and IL‑6 amplify inflammatory cascades by enhancing cytokine production and immune cell differentiation and by promoting osteoclastogenesis and bone resorption.2,3,11 The IL‑23/IL‑17 pathway appears particularly relevant to the coexistence of osteoarticular and cutaneous manifestations, as IL‑17 contributes to neutrophil activation, chronic tissue inflammation, and dysregulated bone remodeling, including impaired osteoblast function.2,3,11 Increased expression of these cytokines has been demonstrated in peripheral blood and inflammatory lesions in SAPHO and related autoinflammatory bone disorders, supporting a biologically plausible rationale for targeted therapy.2,10 These mechanistic insights have guided the off‑label use of bDMARDs targeting TNF, IL‑1, IL‑6, IL‑17, and IL‑23 in patients with refractory disease.10,11 In addition, Janus kinase inhibitors (JAKis) offer a broader therapeutic approach by blocking intracellular signaling of multiple proinflammatory cytokines via the JAK/signal transducer and activator of transcription pathway, which integrates signals from several cytokine families implicated in SAPHO pathophysiology, and may therefore affect both osteoarticular and cutaneous disease domains.3,11
Although targeted therapies have been increasingly reported, the evidence base remains limited to isolated case reports and small case series. No completed RCTs exist, although an RCT evaluating etanercept in SAPHO is currently underway (Study of the efficacy and safety of etanercept treatment in patients with SAPHO syndrome; NCT06011889). Two systematic reviews have been published: the one from 2019 focused mainly on TNFis and did not include IL‑17/IL‑23 inhibitors or JAKis14; and another from 2023 excluded studies with fewer than 5 cases, studies lacking safety outcomes, and those involving patients with isolated CRMO/CNO.11 Given that the modified Kahn criteria9 include nonbacterial osteitis as part of the SAPHO spectrum, such exclusions may substantially limit evidence synthesis in a rare disease.
The objective of this systematic review was to comprehensively evaluate the efficacy of b/tsDMARDs in SAPHO syndrome, including CRMO/CNO and case reports, across clinical and, where available, radiological outcomes, regardless of comparator or outcome definitions.
Patients and methods
This systematic review was conducted in accordance with the PRISMA guidelines,15 with predefined modifications appropriate for evidence derived predominantly from case reports and case series.
Eligibility criteria
We included publications of any design (case reports, case series, cohort studies, and clinical trials) reporting the efficacy of b/tsDMARDs in patients with SAPHO syndrome classified according to the modified Kahn criteria.1 Exclusion criteria were: non‑English language, lack of access to full text, absence of treatment details (drug name or class), and review articles without a new case description. No restrictions were applied regarding follow‑up duration, comparators, or the specific outcome measures used.
Information sources and study selection
A systematic search of PubMed was conducted through March 31, 2025. Search terms included: “SAPHO,” “CRMO,” “CNO,” “treatment,” “TNF inhibitors,” “IL‑17 inhibitors,” “IL‑23 inhibitors,” “IL‑6 inhibitors,” “IL‑1 inhibitors,” “JAK inhibitors,” “biological drugs,” “biologic agents,” and “targeted synthetic drugs.” The full search strategy is provided in Supplementary material. Reference lists of included articles and relevant reviews were screened to identify additional studies.
After removal of duplicates, titles and abstracts were screened to exclude studies not involving b/tsDMARD treatment or lacking full text. Full texts were then assessed for eligibility. Screening was performed independently by JW, AA, and EK; discrepancies were resolved through discussion and consensus.
Data extraction
The extracted data included the number of participants, demographics, cutaneous and skeletal manifestations, follow‑up duration, b/tsDMARD type (specific drug or class), and treatment response for skeletal and cutaneous domains.
As outcome reporting across studies was heterogeneous and largely descriptive, treatment response was categorized as complete response, partial response, no response, or relapse. Classification was based on the description of clinical improvement or resolution of musculoskeletal symptoms (eg, bone pain or arthritis) and / or improvement on imaging when reported (eg, regression of bone marrow edema on magnetic resonance imaging [MRI], decreased tracer uptake on scintigraphy). Cutaneous outcomes were classified according to reported changes in skin lesions, including palmoplantar pustulosis, psoriasis, or acne (eg, reduction or disappearance of lesions). Standardized dermatologic scores were rarely reported in the included studies. Disagreements in outcome classification were resolved by consensus among the reviewers.
To reduce the risk of duplicate patient inclusion, the reports were screened for potential overlap by comparing institution, patient demographics, clinical characteristics, and treatment details. This assessment was performed independently by multiple reviewers, and suspected duplicates were discussed and resolved by consensus.
Risk of bias and quality assessment
Formal assessment of the risk of bias or methodological quality was not performed. The available evidence consisted exclusively of case reports and small case series, for which validated risk‑of‑bias tools (eg, Cochrane RoB, ROBINS‑I) are not applicable and would not provide meaningful or interpretable results. Consequently, items related to risk of bias assessment in the PRISMA checklist were not fulfilled, and the review should be considered a modified PRISMA‑compliant systematic review.
Statistical analysis
For each drug class, we calculated the median follow‑up duration and the proportion (percentage) of cases achieving complete response in both skeletal and skin outcomes. Descriptive statistics were used throughout. Calculations were performed using Statistica software (version 13.3; TIBCO Software Inc., Palo Alto, California, United States).
Results
The database search identified 1598 records, with an additional 17 identified through reference screening. After the removal of duplicates, 1610 records were screened by title and abstract, and 179 full texts were retrieved and assessed for eligibility. Five potentially relevant reports could not be retrieved due to a lack of library access (listed in Supplementary material), and 3 were excluded due to a lack of full text. The final review included 106 studies (listed in Supplementary material) describing 385 cases of b/tsDMARD use in SAPHO. The flow diagram of full systematic literature search results is presented in Figure 1.

No results of RCTs were found. A single post hoc analysis of a phase 3 randomized, double‑blind, placebo‑controlled trial of guselkumab in Japanese patients with palmoplantar pustulosis included a subgroup with pustulotic arthro‑osteitis fulfilling SAPHO criteria. All remaining studies were case reports or case series. The results of the review are synthetized in Figure 2.

Abbreviations: b/tsDMARD, biologic and targeted synthetic disease‑modifying antirheumatic drug; IL, interleukin; JAKi, Janus kinase inhibitor; PDE4i, phosphodiesterase‑4 inhibitor; SAPHO, synovitis, acne, pustulosis, hyperostosis, and osteitis; TNFi, tumor necrosis factor inhibitor
Demographic data were inconsistently reported across studies. Summary statistics were calculated only for patients with available age and sex information. In the patients with available information, the median (interquartile range [IQR]) age was 41 (27–49) years for TNFis, 38 (29.5–45.5) years for JAKis, 39 (32.5–49.8) years for IL‑17 inhibitors (IL‑17is), 43 (35–54) years for IL‑23is, and 48 (39.5–53) years for other targeted therapies. Women predominated in most treatment groups (56.8%, 70%, 71.4%, and 87.5% respectively), except for the small group receiving other targeted therapies (45.5%).
Both adult and pediatric patients were represented, reflecting the inclusion of CRMO/CNO within the SAPHO spectrum. Pediatric patients were rarely represented in the included reports. Based on available age data, 10 cases involved patients younger than 18 years. Most pediatric cases were treated with TNFis (n = 7). Two pediatric cases treated with a JAKi and 1 case treated with an IL‑1i were also identified. Due to the small number of pediatric cases and heterogeneous reporting, no separate analysis of treatment outcomes in children was feasible.
Tumor necrosis factor inhibitors
A total of 77 studies described TNFi treatment in 203 patients (Supplementary material, Table S1). Etanercept was used in 61 patients, adalimumab in 54, infliximab in 42, certolizumab and golimumab each in 2, and the TNFi was not specified in 42 cases. Median follow‑up duration was 9 (3–24) months. Complete skeletal response was achieved in 157/195 patients (81%), and complete skin response in 112/156 (72%).
Janus kinase inhibitors
Thirty‑three studies reported on JAKi treatment in 77 patients (Supplementary material, Table S2). Tofacitinib was used in 54 patients, baricitinib in 9, upadacitinib in 2, and abrocitinib in 1, with the specific agent not provided in 11 cases. Median follow‑up was 3 (3–10) months. Complete skeletal response occurred in 51/57 patients (89%), and complete skin response in 49/52 (94%).
Interleukin 17 inhibitors
Fifteen studies described IL‑17i treatment in 31 patients (Supplementary material, Table S3). Secukinumab was used in 28 cases, ixekizumab in 1, and brodalumab in 2. Median follow‑up was 4 (1.5–6) months. Complete skeletal response was observed in 20/31 patients (65%), and complete skin response in 12/29 (41%).
Interleukin 23 inhibitors
Ten studies reported IL‑23i treatment in 56 patients (Supplementary material, Table S4). Guselkumab was used in 45 patients, ustekinumab in 8, risankizumab in 2, and tildrakizumab in 1. Median follow‑up was 12 (12–12) months, with most cases reported at 12‑month follow‑up. Complete skeletal response was achieved in 33/56 patients (59%), and complete skin response in 8/10 (80%).
A separate exploratory analysis from the guselkumab RCT subgroup (45 patients with pustulotic arthro‑osteitis meeting the modified Kahn criteria) showed improvements at week 52 in skeletal pain (58.6%).16 The mean change in palmoplantar psoriasis area and severity index was 23.6.
Other biologic and targeted synthetic disease‑modifying antirheumatic drugs
Reports on other targeted therapies were limited to single cases or small series (Supplementary material, Table S5). Four studies described IL‑1i use, with anakinra administered to 9 patients, and median follow‑up of 5 (4–8) months, during which complete skeletal response was observed in 6 patients and complete skin response in all 3 patients with cutaneous involvement. Three studies reported treatment with the IL‑6i tocilizumab in 5 patients, with median follow‑up of 4.5 (1–8.5) months; 3 patients achieved a complete skeletal response, while none of the 3 patients with skin manifestations experienced complete resolution. Additionally, 4 studies documented the use of the phosphodiesterase‑4 inhibitor apremilast in 4 patients, with median follow‑up of 7 (5–12.5) months, complete skeletal response in 2 of 4 evaluable cases, and complete skin response in 2 of 3 patients.
Radiological outcomes
Radiological outcomes were reported inconsistently and only in a minority of the included studies; therefore, imaging findings are summarized separately from clinical outcomes. Imaging modalities included MRI, positron emission tomography / computed tomography (PET/CT), CT, and bone scintigraphy, most commonly for assessment of osteitis or bone marrow edema.
Radiological responses in the patients receiving TNFis were variable and did not always parallel clinical improvement. Cases of improvement visible on MRI or PET/CT were reported with adalimumab and etanercept, whereas persistent inflammatory activity despite clinical response was also described in infliximab‑treated patients.17-20 For IL‑17 inhibition, MRI improvement or remission of osteitis was reported in most evaluable cases treated with secukinumab, although partial responses were also observed.21,22 Radiological outcomes were more frequently described in reports of JAKi therapy. MRI improvement of inflammatory bone lesions was seen in several case reports and in a small prospective study in which most patients demonstrated partial MRI improvement, including some with discordant clinical and imaging responses.23-25 For IL‑23is, imaging data were available primarily from an exploratory analysis of guselkumab in pustulotic arthro‑osteitis, showing MRI improvement in inflammatory lesions at week 52 in 43% of the patients.16 A patient treated with ustekinumab demonstrated partial MRI improvement.23 Among other targeted therapies, imaging outcomes were sporadically reported. Improvement on scintigraphy or MRI was described with anakinra, whereas both partial MRI improvement and radiological progression were reported in patients treated with tocilizumab.18,26-29
Overall, the limited and heterogeneous reporting of imaging outcomes precluded their quantitative synthesis.
Discussion
This systematic review, encompassing 106 studies, highlights the growing use of b/tsDMARDs in the treatment of SAPHO syndrome. The 2019 systematic review identified only 66 cases of patients treated with bDMARDs.14 Our systematic review describes 385 cases treated with b/tsDMARDs. As the earliest biologic class introduced in rheumatology, TNFis are the most frequently reported in SAPHO. In the absence of contraindications, they appear to be a reasonable first‑line biologic option for patients who do not respond to conventional therapies. The ongoing RCT evaluating etanercept may help substantiate this approach and provide much‑needed evidence‑based guidance.
JAKis represent the second most extensively documented therapeutic class, and have shown high complete response rates in available case reports and series. Their broad mechanism of action—blocking intracellular signaling downstream of multiple proinflammatory cytokines—may be advantageous in a disease as immunologically complex as SAPHO.2,3,11 In contrast, the inhibitors targeting individual cytokines (IL‑1, IL‑6, IL‑17, or IL‑23) showed more variable efficacy. This heterogeneity of response likely reflects the multifactorial cytokine network underlying SAPHO pathogenesis rather than the dominance of a single inflammatory pathway.2,3,10 Nevertheless, given anecdotal evidence of partial benefit, these agents may serve as reasonable alternatives in cases of inadequate response, intolerance, or contraindications to TNF or JAK inhibition.
Recently published international consensus recommendations have proposed the term adult CNO and outlined a treatment approach based primarily on NSAIDs, TNFis, and bisphosphonates.9 However, these recommendations are largely based on expert consensus and limited clinical evidence. The present review complements the recommendations by summarizing the accumulated case‑based experience with b/tsDMARDs reported in patients historically classified as having SAPHO syndrome. These data highlight that, in the patients with refractory or chronic disease, clinicians have explored a broader range of targeted therapies than those currently emphasized in the consensus recommendations. Importantly, many cases included in our review presented with a broader SAPHO phenotype, including cutaneous manifestations, indicating that the SAPHO concept remains clinically relevant for a substantial proportion of reported patients.
An important observation from this review is the limited and inconsistent reporting of radiological outcomes. Although imaging improvement of osteitis or bone marrow edema was described across several therapeutic classes, these data were available only for a small subset of cases and were assessed using heterogeneous modalities. In several reports, clinical improvement did not correspond with imaging findings, highlighting the complexity of disease assessment in SAPHO syndrome. The significance of radiological response as a measure of treatment effectiveness remains unclear. These observations underscore the need for standardized radiological outcome measures in future studies.
Limitations
The main limitation of this review is the low quality of the available evidence. All included studies were case reports or case series, reflecting the rarity of SAPHO syndrome and the absence of acute life‑threatening manifestations, which likely limit prioritization of this condition for large, commercially sponsored clinical trials. Another important limitation is the substantial heterogeneity of the included reports. Patient characteristics, disease manifestations, outcome definitions, and follow‑up duration varied widely across studies. As a result, comparisons between different drug classes should be interpreted with caution. The response rates reported in this review represent a descriptive aggregation of published cases rather than a formal comparative effectiveness analysis. In addition, median follow‑up differed markedly between therapeutic classes (eg, shorter for JAKis than for TNFis), which may influence the apparent response rates and further limit direct cross‑drug comparisons. In addition, publication bias is likely substantial, as successful treatment experiences are more readily reported. These limitations underscore the urgent need for higher‑quality evidence, ideally through randomized trials or prospective registries that employ standardized outcome measures.
Another limitation of our review is that the literature search was limited to a single database. We attempted to mitigate this by screening reference lists of the included publications, though some degree of omission remains possible. Additionally, although we sought full texts from study authors, 5 potentially eligible reports could not be obtained. Finally, because the review is based largely on case reports and small case series, the possibility of duplicate patient reporting across publications cannot be completely excluded despite manual screening for overlapping cases.
Conclusions
This systematic review provides a consolidated overview of available evidence on b/tsDMARD use in SAPHO syndrome, a condition for which no treatment guidelines currently exist. Although the evidence base is limited to low‑quality, uncontrolled studies, the accumulated data suggest that TNFis and JAKis are the most consistently effective options in the patients who do not respond to conventional therapy. Robust clinical evidence—ideally from RCTs or prospective registries—is urgently needed to inform evidence‑based management of this rare disease.
- Zimmermann P, Curtis N. Synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO) syndrome ‑ a challenging diagnosis not to be missed. J Infect. 2016; 72: S106‑S114. | Crossref
- Yang Y, Chen Q, Zhong W. The role of cytokines in the pathogenesis of SAPHO syndrome. Front Immunol. 2024; 15: 1427784. | Crossref
- Ferraioli M, Levani J, De Luca R, et al. What is new and what is next for SAPHO syndrome management: a narrative review. J Clin Med. 2025; 14: 1366. | Crossref
- Cao Y, Li C, Xu W, et al. Spinal and sacroiliac involvement in SAPHO syndrome: a single center study of a cohort of 354 patients. Semin Arthritis Rheum. 2019; 48: 990‑996. | Crossref
- Hayem G, Bouchaud‑Chabot A, Benali K, et al. SAPHO syndrome: a long‑term follow‑up study of 120 cases. Semin Arthritis Rheum. 1999; 29: 159‑171. | Crossref
SUPPLEMENTARY MATERIAL
ARTICLE INFORMATION