Pulmonary carcinoids are rare neuroendocrine neoplasms characterized by generally indolent behavior with a recognized potential for late recurrence.
According to the 2021 World Health Organization classification,1 pulmonary neuroendocrine tumors include typical and atypical carcinoids, which differ in biological aggressiveness and prognosis. Typical carcinoids are low‑grade neoplasms characterized by indolent growth and a relatively low risk of recurrence (3%–6%) following complete resection. Most relapses occur within the first 5 years, while late recurrences are usually reported between 10 and 15 years. Recurrences beyond 25 years are exceedingly rare.
Current guidelines2 recommend long‑term surveillance due to the potential for late relapse. Computed tomography (CT) imaging is typically performed every 6–12 months during the first few years, and less frequently thereafter. However, no fixed end point for follow‑up has been established, and surveillance rarely extends beyond 10–15 years in clinical practice. Long‑term follow‑up poses a clinical challenge, requiring a balance between surveillance benefits and the risk of cumulative radiation exposure and secondary malignancies.3
We present a case of a 57‑year‑old woman who underwent segmentectomy of segment 6 of the right lung in 1990 for a typical carcinoid tumor. Postsurgery, she remained asymptomatic for many years. In January 2025, she presented with a persistent cough. Imaging showed a mass in the right lung measuring 43 mm × 33 mm × 35 mm, extending into the lower lobe bronchus and causing airway compression (Figure 1A).
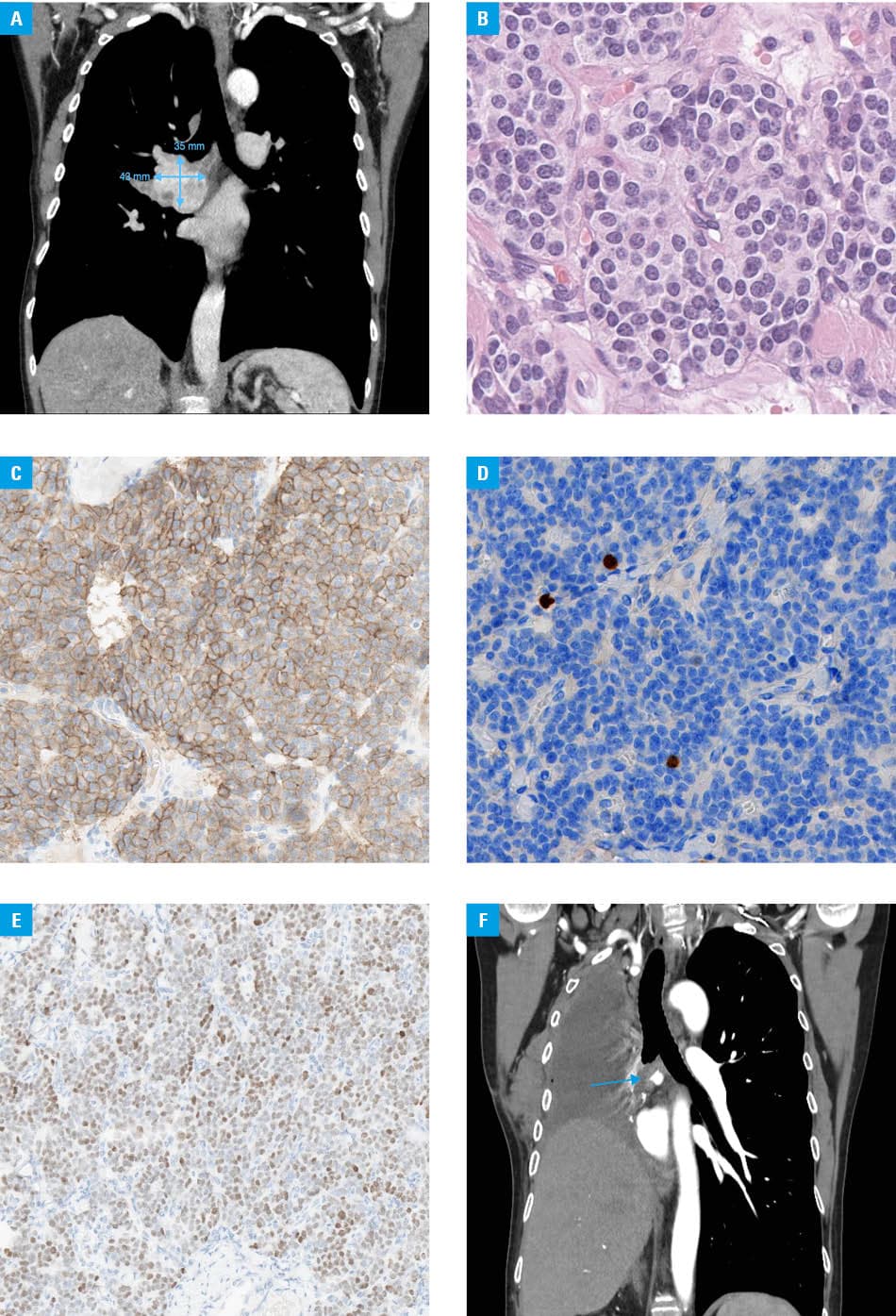
Bronchoscopic biopsy confirmed a neuroectodermal tumor with positive staining for CD56, synaptophysin, and chromogranin. The Ki67 index was approximately 1%. [68Ga]Ga‑DOTA‑0‑Tyr3‑Octreotate positron emission tomography/CT scan showed somatostatin receptor expression in the primary tumor, regional lymph nodes, and a solitary pelvic bone lesion. Based on tumor board assessment, the pelvic lesion was considered metastatic; biopsy was deemed unnecessary given its imaging characteristics and therapeutic implications. Treatment with a long‑acting somatostatin analog was initiated. Given the potential for radical management, stereotactic radiotherapy (27 Gy) to the bone lesion was performed, followed by right‑sided pneumonectomy with mediastinal lymph node dissection in December 2025. Histopathological examination confirmed a typical carcinoid tumor (G1; Ki67 index, 1%; mitotic index, 0–1 mitoses/2 mm2; no necrosis; Figure 1B–1E) with metastasis in 1/45 lymph nodes and R0 resection.
Postoperative imaging (Figure 1F) showed a soft‑tissue lesion near the bronchial stump, initially considered postoperative; further evaluation is ongoing. Treatment with a long‑acting somatostatin analog is continued. Although the presentation suggests an ultra‑late recurrence, a second metachronous primary neoplasm cannot be excluded due to a lack of archival tissue for comparison. Genetic testing excluded multiple endocrine neoplasia type 1 syndrome, suggesting a sporadic origin.
Very few reports have described recurrences 25–30 years after surgery,4-6 and, to the best of our knowledge, this is among the latest reported recurrences of a typical pulmonary carcinoid after curative resection, and may represent the longest disease‑free interval reported in the contemporary literature. Notably, recurrence was detected due to symptoms rather than routine surveillance.
These findings challenge the assumption that long disease‑free survival is tantamount to being cured and suggest that follow‑up strategies may underestimate the ultra‑late recurrence risk, even in tumors with a very low Ki67 index. Indefinite routine imaging for all patients is not feasible, requiring a balance between surveillance benefits and radiation‑related risks of cumulative radiation exposure.
- WHO Classification of Tumours Editorial Board Thoracic Tumours. WHO classification of tumours series. 5th ed Volume 5. International Agency for Research on Cancer. 2021. https://publications.iarc.fr/595. Accessed March 20, 2026. | Crossref
- Singh S, Bergsland EK, Card CM, et al. Commonwealth Neuroendocrine Tumour Research Collaboration and the North American Neuroendocrine Tumor Society Guidelines for the Diagnosis and Management of Patients with Lung Neuroendocrine Tumors: An International Collaborative Endorsement and Update of the 2015 European Neuroendocrine Tumor Society Expert Consensus Guidelines. J Thorac Oncol. 2020; 15: 1577‑1598. | Crossref
- Heijboer FWJ, Mulders TA, van Straten M, et al. Radiological follow‑up in patients with resected pulmonary carcinoids: should we reduce radiation exposure? Lung Cancer. 2024; 198: 108030. | Crossref
- Bełz A, Rosiek V, Głogowska‑Szeląg J, et al. Lung nodule 25 years after lobectomy ‑ recurrence of bronchial carcinoid. Wiad Lek. 2020; 73: 23092312. | Crossref
- Townshend AP, Lakshminarayanan B, Ucar AE, et al. Rare pleural recurrence of typical pulmonary carcinoid tumor 30 years after lobectomy. Ann Thorac Surg. 2007; 83: 15231524. | Crossref
ARTICLE INFORMATION