Congenital adrenal hyperplasia (CAH) comprises a group of genetic disorders requiring lifelong glucocorticoid replacement therapy to suppress excessive adrenocorticotropic hormone (ACTH) secretion. Untreated disease causes chronic cortisol deficiency and secondary ACTH overproduction.1 Chronic ACTH level elevation drives adrenal hyperplasia and, in some cases, leads to the occurrence of masses, including myelolipomas—benign tumors composed of fat and hematopoietic tissue.2 Imaging findings may mimic adrenal incidentalomas or malignant tumors.
This case demonstrates a diagnostic pitfall in untreated CAH, where an exceptionally large adrenal mass suggestive of myelolipoma or adrenocortical carcinoma proved to be an adenoma with fatty metaplasia on histology.
A 37‑year‑old woman with salt‑wasting CAH diagnosed in the neonatal period, off treatment since the age of 18 years, was admitted after trauma. Computed tomography showed a cystic‑solid left adrenal mass (119 mm × 92 mm × 67 mm) with nodular right adrenal enlargement, initially suggestive of cancer. The patient was referred to the Department of Endocrinology, Diabetology, and Internal Medicine at the Medical University of Bialystok, Poland for further evaluation.
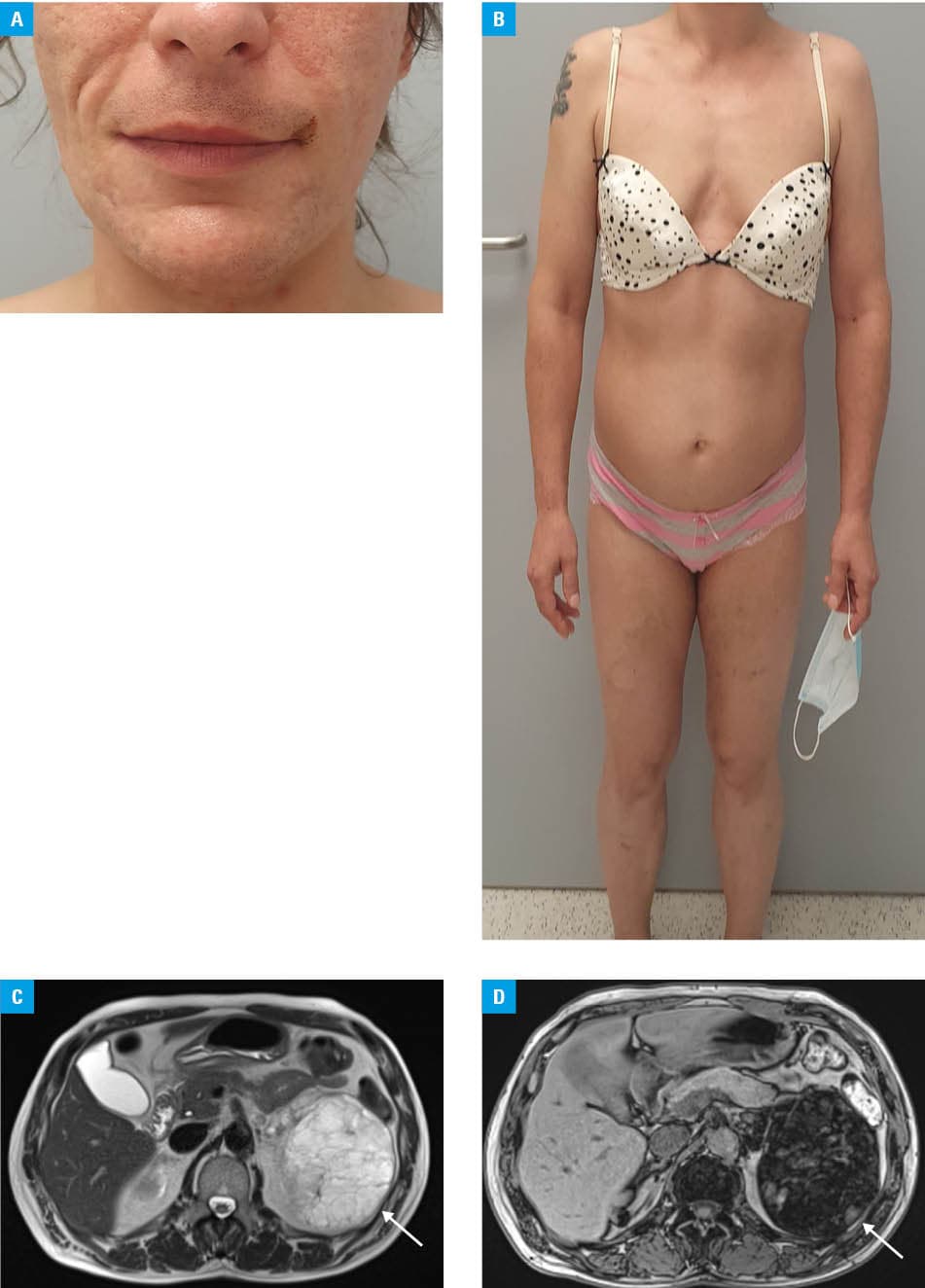
Physical examination showed generalized hirsutism and virilization (Figure 1A and 1B). Laboratory test results showed elevated levels of 17‑hydroxyprogesterone, testosterone, and ACTH, with decreased levels of cortisol and estradiol, dehydroepiandrosterone sulfate (DHEA‑S) level within the reference range, and an increased level of sex hormone–binding globulin (SHBG; Supplementary material, Table S1). Graves disease was also diagnosed. The diagnosis of salt‑wasting CAH due to 21‑hydroxylase deficiency, established in the neonatal period, was reconfirmed biochemically.3
Although the DHEA‑S level was within the reference range, sustained ACTH‑driven adrenal steroidogenesis could not be excluded, as androgen production in 21‑hydroxylase deficiency also occurs through backdoor and 11‑oxygenated pathways that are not adequately reflected by the DHEA‑S concentration alone.4 The SHBG level elevation in our patient, atypical of hyperandrogenism (in which elevated androgens usually suppress SHBG), is better explained by the coexisting thyrotoxicosis, which increases hepatic SHBG synthesis, and argues against an autonomous androgen‑secreting tumor.5
The lipid‑rich adenoma, myelolipoma, and adrenocortical carcinoma considered here are differentiated by unenhanced attenuation and contrast washout.1,2 Since they could not be assessed (scan performed externally, medical records inaccessible), magnetic resonance imaging was performed, showing a large polycyclic left adrenal mass with an opposed‑phase signal drop consistent with intracellular lipid component, together with separate lipid‑containing nodules in the contralateral right adrenal gland (Figure 1C and 1D). The imaging findings were most consistent with myelolipoma, but lipid‑rich adenoma or carcinoma could not be excluded.
Due to the inability to exclude carcinoma and considerable lesion size, adrenalectomy was indicated.1,2 Since the tumor was large and potentially malignant in nature, an open approach was chosen over laparoscopy to enable intact removal without capsular rupture.2 Histologic examination showed an adrenocortical adenoma (inhibin- and pan‑cytokeratin–positive), with fatty metaplasia and lymphocytic infiltration of uncertain significance. Adrenocortical adenomas may rarely undergo fatty (lipomatous) metaplasia with accumulation of mature adipose tissue within the tumor. The resulting macroscopic fat can mimic the imaging appearance of myelolipoma, whereas the intracellular lipid component may still produce opposed‑phase signal loss characteristic of a lipid‑rich adenoma, thereby explaining the discordant imaging features.2
Levels of laboratory parameters normalized after initiation of treatment with fludrocortisone, dexamethasone, and methimazole; except cortisol, whose level remains low in individuals receiving dexamethasone, which does not indicate undertreatment.
Adrenal masses in patients with untreated CAH require individualized assessment.1,2 Uncomplicated hyperplasia does not require surgery; however, our patient presented with a large, indeterminate mass, which prompted surgical treatment, followed by hormone replacement, monitoring, treatment of Graves disease, and surveillance of the remaining adrenal gland.1,3
- Nermoen I, Falhammar H. Prevalence and characteristics of adrenal tumors and myelolipomas in congenital adrenal hyperplasia: a systematic review and meta‑analysis. Endocr Pract. 2020; 26: 1351‑1365. | Crossref
- Calissendorff J, Juhlin CC, Sundin A, et al. Adrenal myelolipomas. Lancet Diabetes Endocrinol. 2021; 9: 767‑775. | Crossref
- Speiser PW, Arlt W, Auchus RJ, et al. Congenital adrenal hyperplasia due to steroid 21‑hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2018; 103: 4043‑4088. | Crossref
- Yang M, White PC. Genetics and pathophysiology of classic congenital adrenal hyperplasia due to 21‑hydroxylase deficiency. J Clin Endocrinol Metab. 2025; 110: S1‑S12. | Crossref
- Kjaergaard AD, Marouli E, Papadopoulou A, et al. Thyroid function, sex hormones and sexual function: a Mendelian randomization study. Eur J Epidemiol. 2021; 36: 335‑344. | Crossref
SUPPLEMENTARY MATERIAL
ARTICLE INFORMATION