Ectopic acromegaly due to growth hormone–releasing hormone secretion from bronchial carcinoid causing somatotroph hyperplasia and partial pituitary insufficiency



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Ectopic acromegaly due to growth hormone–releasing hormone secretion from bronchial carcinoid causing somatotroph hyperplasia and partial pituitary insufficiency
Acromegaly due to ectopic growth hormone–releasing hormone (GHRH) secretion from a neuroendocrine tumor (NET) is very rare, and up to 100 cases have been reported in the literature.1 Pancreatic or bronchial NETs are the primary sources of GHRH, but pheochromocytomas were also described.2-5 To the best of our knowledge, we report the first case of acromegaly due to GHRH‑producing NET causing pituitary hyperplasia and resulting in partial pituitary insufficiency.
A 43‑year‑old woman was referred to our department with symptoms suggesting acromegaly for about 5 years and amenorrhea for 2 years. The patient presented with typical acromegaly symptoms: coarsened facial features, macroglossia, enlarged hands and feet, soft tissue swelling, marked interdental spacing, and excessive sweating (Figure 1A). Her medical history was notable for bilateral surgery for carpal tunnel syndrome. Hormonal evaluation revealed normal thyroid function, normal prolactin levels, hypogonadotropic hypogonadism (luteinizing hormone [LH], 1.3 U/l; follicle‑stimulating hormone [FSH] 6.3 U/l, and estradiol <10 pg/ml), secondary hypocortisolism (adrenocorticotropic hormone [ACTH] 08:00 AM, 6.2 pg/ml; cortisol 8:00 AM, 3.5 µg/dl), elevated fasting growth hormone (GH) levels (44 µg/l), and insulin‑like growth factor 1 (IGF‑1) levels exceeding 3.3‑fold the upper limit of normal (ULN). Nonsuppressed GH levels during the 75‑g oral glucose tolerance test were noted (nadir, 17 µg/l).
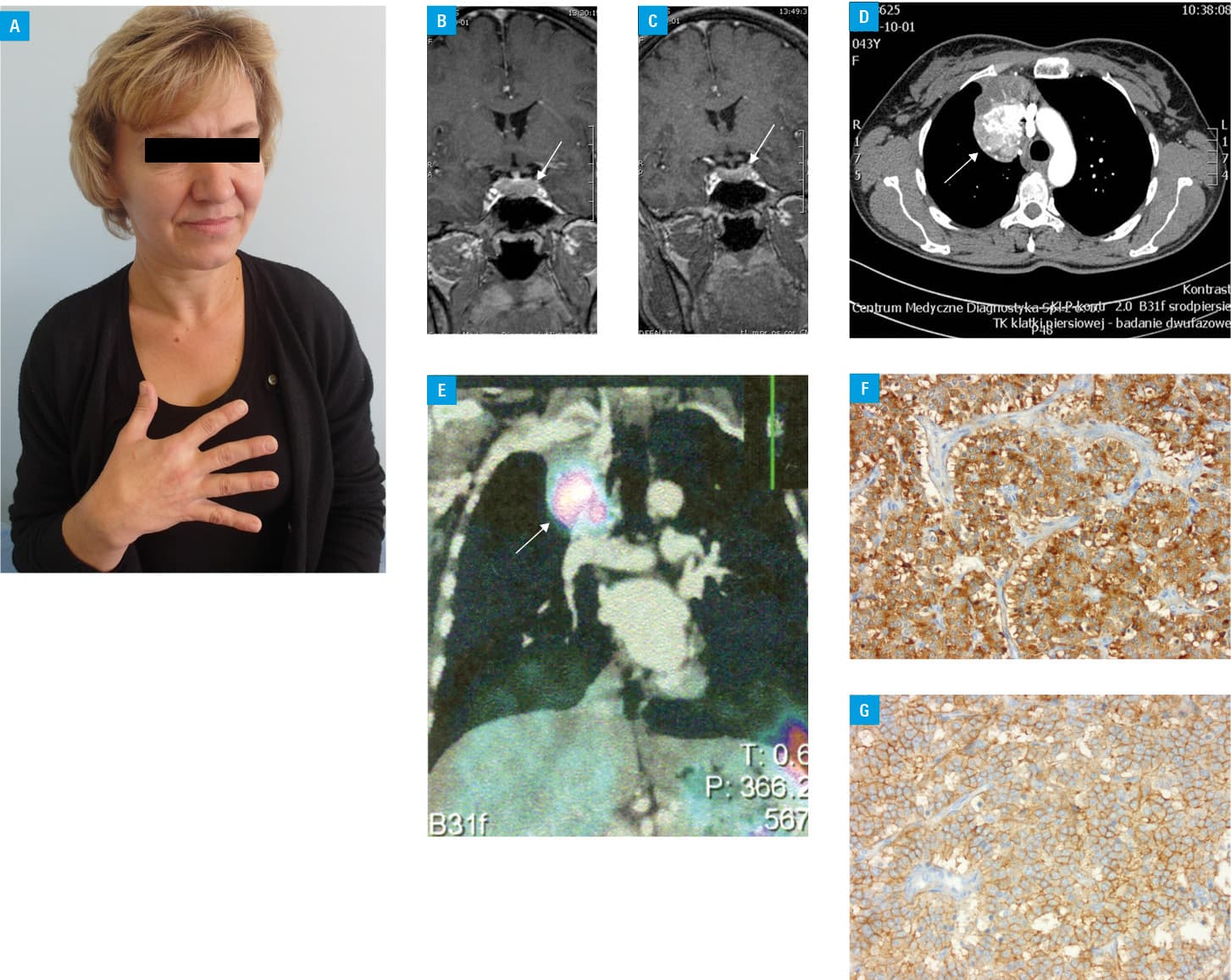
Pituitary magnetic resonance imaging revealed an enlarged gland (24 × 13 × 12 mm), with extrasellar extension, and homogenous gadolinium enhancement without focal lesion (Figure 1B). As no pituitary adenoma could be detected, a chest X‑ray was performed. A tumor (56 × 40 mm) in the right anterior mediastinum was identified. Chest computed tomography (CT) confirmed the presence of a 5‑cm tumor with calcifications and strong contrast enhancement (Figure 1C). Somatostatin receptor scintigraphy showed abnormal radiolabel uptake of the tumor revealed by CT (Figure 1D). Long‑acting somatostatin analogue treatment with lanreotide Autogel (120 mg) was started while awaiting surgery. A significant improvement in acromegaly symptoms was observed. After 3 months of treatment, pituitary imaging showed a reduction in the pituitary size (19 × 13 × 8 mm) (Figure 1E) associated by a decrease in GH and IGF‑1 levels (GH, 6 µg/l; IGF‑1, 1.4 × ULN), normalization of corticotroph function (ACTH 8:00 AM, 14 pg/ml; cortisol 8:00 AM, 9.4 µg/dl) and gonadotrophic function (LH, 8.9 U/l; FSH, 7.9 U/l; estradiol, 177 pg/ml) with regular menstrual cycles. No reduction in the pulmonary tumor size on CT was noted. The patient underwent a right upper lobectomy with clear tumor margins. A pathological report revealed a typical carcinoid with a mitotic count of less than 2 mitoses/2 mm2 and absence of necrosis. Immunostaining was positive for chromogranin and CD56. Additional staining of the tumor showed high expression of GHRH and SSTR2 (80% of the cells) (Figure 1F and 1G). The concentrations of GH and IGF‑1 normalized after surgery (GH, 0.57 µg/l; IGF‑1, 0.97 × ULN). No recurrence of acromegaly symptoms during a 3‑year follow‑up was observed.
In summary, we reported a case of acromegaly with transient pituitary insufficiency due to GHRH‑producing bronchial carcinoid causing somatotroph hyperplasia. A distinction between a pituitary somatotroph adenoma and ectopic GHRH secretion is important, as pituitary hyperplasia may be misdiagnosed as a pituitary tumor, leading to unnecessary pituitary surgery.2 Long‑standing pituitary hyperplasia may lead to a deficiency in one or more pituitary hormones.
- Ghazi AA, Amirbaigloo A, Dezfooli AA, et al. Ectopic acromegaly due to growth hormone releasing hormone. Endocrine. 2013; 43: 293‑302. | Crossref
- Garby L, Caron P, Claustrat F, et al. Clinical characteristics and outcome of acromegaly induced by ectopic secretion of growth hormone‑releasing hormone (GHRH): a French nationwide series of 21 cases. J Clin Endocrinol Metab. 2012; 97: 2093‑2104. | Crossref
- Butler PW, Cochran CS, Merino MJ, et al. Ectopic growth hormone‑releasing hormone secretion by a bronchial tumor: clinical experience following tumor resection and long‑acting octreotide therapy. Pituitary. 2012: 15: 260‑265.
- Mumby C, Davis JR, Trouillas J, Higham C. Pheochromocytoma and acromegaly. Endocrinol Diabetes Metab Case Rep. 2014; 2014: 140036.
- Bolanowski M, Schopohl J, Marciniak M, et al. Acromegaly due to large bronchial carcinoid. Complete recovery following tumor surgery. Exp Clin Endocrinol Diabetes. 2002; 110: 188‑192. | Crossref
ARTICLE INFORMATION