Roxadustat: another drug that causes pulmonary hypertension? Report of first human case



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Roxadustat: another drug that causes pulmonary hypertension? Report of first human case
Roxadustat (INN, FG‑4592) is a first‑in‑class oral drug for treating anemia in chronic kidney disease. It is a second‑generation hypoxia‑inducible factor (HIF) prolyl‑hydroxylase‑2 (PHD2) inhibitor.1 The drug proved effective in recent clinical trials (phase III OLYMPUS and ROCKIES trials for roxadustat met their primary endpoints in chronic kidney disease patients with anemia) and was recently granted market approval in China. We present a patient who developed severe pulmonary arterial hypertension (PAH) while being treated in a phase III clinical trial of roxadustat. Such an association has never been described for a PHD2 inhibitor before. HIF-α hydroxylase system is involved in the regulation of vascular remodeling.
A 74‑year‑old woman was hospitalized due to sudden unexplained worsening of exercise tolerance and shortness of breath. Her medical history included stable chronic angina (remote myocardial infarction without detectable left ventricular dysfunction), hypothyroidism, and diabetes. In May 2016, she was recruited for phase III trial with oral roxadustat in adjusted doses of 40 to 100 mg 3 times a week because of her stage G4 chronic kidney disease with anemia. The trial lasted 24 months. After that she was admitted to our department with class III heart failure with the 6‑minute walk test distance of 100 m. An electrocardiogram showed sinus rhythm at 70 bpm with flat negative T waves in all leads. Laboratory tests on admission showed a glomerular filtration rate of 20.5 ml/min/1.73 m2, hemoglobin level of 10.6 g/dl, and N‑terminal fragment of the prohormone brain natriuretic peptide (NT‑proBNP) level of 14596 pg/ml. Echocardiography showed enlargement of the right cavities of the heart, features of PAH with depressed right ventricular systolic function (estimated systolic pulmonary artery pressure [PAP], 87 mm Hg; mean PAP, 47 mm Hg; diastolic PAP, 24 mm Hg), and moderate tricuspid regurgitation, whereas left ventricular ejection fraction was normal at 56% (Figure 1). Pulmonary hypertension was not suspected on echocardiography performed in June 2012. Right heart catheterization confirmed irreversible precapillary PAH (mean PAP, 48 mm Hg, pulmonary vascular resistance, 12.3 WU). Pulmonary embolism was excluded.
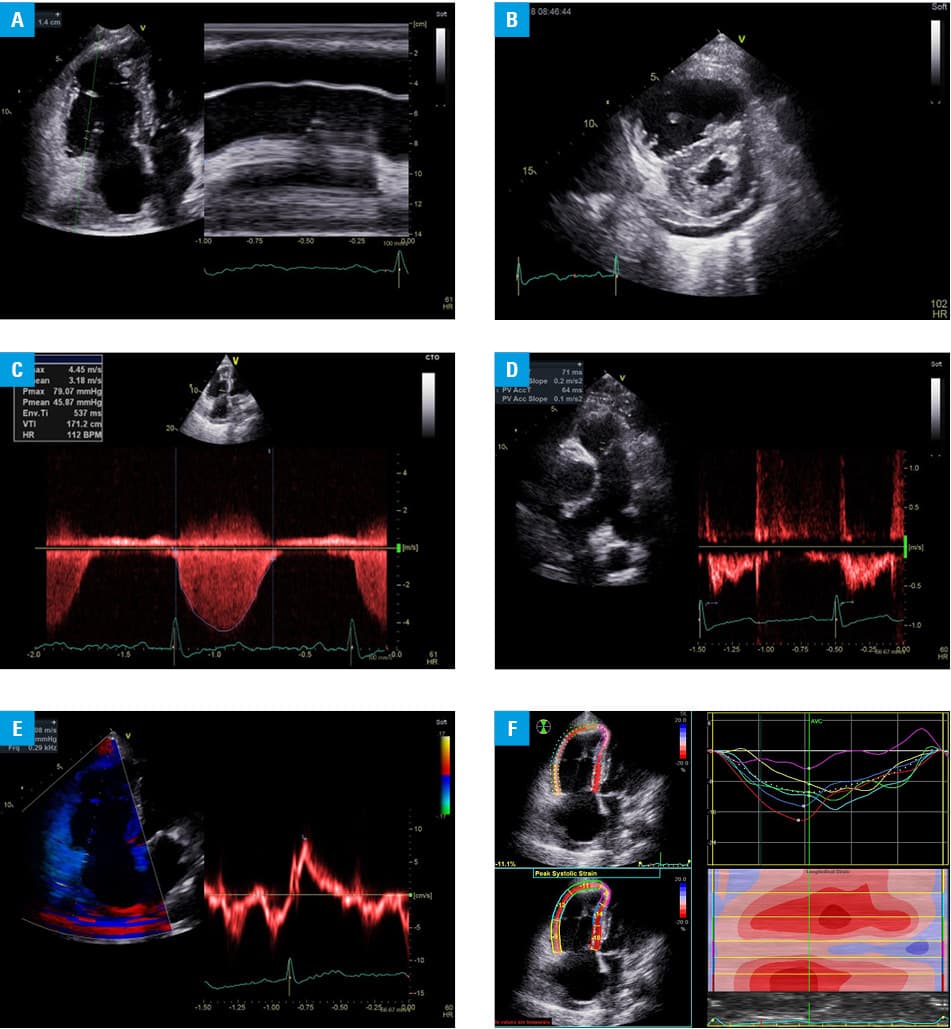
The treatment of PAH was initiated with oral sildenafil (20 mg 3 times a day), and subcutaneous treprostinil, escalated to a current dose of 42.5 ng/kg/min along with escalation of loop diuretics. As roxadustat was considered a potential contributor to the development of PAH, it was discontinued (May 2018) and replaced with erythropoietin. On the last follow‑up on November 19, 2018, after 5 months of PAH‑specific treatment, the 6‑minute walk test distance was 200 m, NT‑proBNP was 1385 pg/ml, and hemoglobin level was 11.2 g/dl. Echocardiographic parameters also improved. Thus, functional improvement was seen even though anemia worsened after withdrawal of roxadustat.
To our best knowledge, this report is probably the first to document the development of severe PAH during roxadustat therapy for anemia. The drug inhibits PHD2, an enzyme responsible for breaking down HIF under normoxic conditions. Stabilization of HIF2α upregulates Notch3 and transforming growth factor β, as well as increases the pericyte coverage and transformation of pericyte into myofibroblasts or vascular smooth muscle cells, which is associated with remodeling of pulmonary arterioles and PAH development.2 Inhibition (or gene knockout) of PHD2 increases the HIF2α level and enhances endothelial‑to‑mesenchymal transition with upregulation of SNAI1/2 genes in the lung. Thus, the HIF pathway is linked with adverse vascular remodeling and experimentally confirmed to induce PAH.3 Roxadustat's mode of action is related to erythropoietin, which was shown to stimulate proliferation of endothelial and smooth muscle cells in pulmonary vasculature.4
We understand that our report cannot prove a causal relationship between roxadustat and development of PAH; however, it is plausible that the mode of action of roxadustat is linked to the pathophysiology of PAH.
- Del Vecchio L, Locatelli F. Roxadustat in the treatment of anaemia in chronic kidney disease. Expert Opin Investig Drugs. 2018; 27: 125‑133. | Crossref
- Wang S, Zeng H, Xie XJ, et al. Loss of prolyl hydroxylase domain protein 2 in vascular endothelium increases pericyte coverage and promotes pulmonary arterial remodeling. Oncotarget. 2016; 7: 58848‑58861. | Crossref
- Tang H, Babicheva A, McDermott KM, et al. Endothelial HIF‑2α contributes to severe pulmonary hypertension due to endothelial‑to‑mesenchymal transition. Am J Physiol Lung Cell Mol Physiol. 2018; 314: L256‑L275.
- Karamanian VA, Harhay M, Grant GR, et al. Erythropoietin upregulation in pulmonary arterial hypertension. Pulm Circ. 2014; 4: 269‑279. | Crossref
ARTICLE INFORMATION