Genetic heterogeneity of indeterminate thyroid nodules assessed preoperatively with next-generation sequencing reflects the diversity of the final histopathologic diagnosis
Key words: follicular thyroid adenoma, follicular thyroid cancer, genetics, next-generation sequencing, whole-genome studies



 CC BY-NC-SA 4.0
CC BY-NC-SA 4.0
Genetic heterogeneity of indeterminate thyroid nodules assessed preoperatively with next-generation sequencing reflects the diversity of the final histopathologic diagnosis
Introduction: Inconclusive cytologic results of thyroid fine‑needle aspiration biopsy (FNAB) include atypia or follicular lesion of undetermined significance (FLUS) and follicular neoplasm or suspicious for follicular neoplasm (SFN).
Objectives: We aimed to assess the genetic background of indeterminate thyroid nodules and to identify new genetic pathways potentially involved in the development of follicular thyroid cancer.
Patients and methods: Genomic DNA was isolated from FNAB samples from 25 white patients (2 men; 23 women) diagnosed preoperatively with FLUS (n = 16) and SFN (n = 9). Next‑generation sequencing (NGS) was performed. The results were compared with clinical data, including final postsurgical diagnoses.
Results: The malignancy rate was 28%. KDR, RET, and TP53 gene mutations were most frequent in FLUS and SFN samples finally diagnosed as cancers, whereas alterations in RET, TP53, FLT3, APC, and PDGFRA predominated in benign tumors. KDR tended to be more common in malignant samples (75% vs 20%, P = 0.1). A total number of mutated genes was higher in patients with benign tumors (17 vs 11, P = 0.02), but there was no difference between groups in the mean number of mutations per patient (4.9 [range, 1–9]).
Conclusions: We showed that the heterogeneity in the genetic background of indeterminate thyroid nodules corresponds to their histopathologic diversity. The role of KDR as a possible malignancy marker needs to be confirmed. Glass slides with FNAB samples may constitute a reliable source of genetic material for NGS studies, providing a better insight into the molecular profile of thyroid nodules.
What's new?
Despite developed diagnostic methods for thyroid nodules, fine‑needle aspiration biopsy (FNAB) may still provide inconclusive results. This is the first study evaluating a wide panel of potential malignancy markers in material obtained from glass slides with FNAB specimens. We showed that the heterogeneity in the genetic background of indeterminate thyroid nodules corresponds to their histopathologic diversity. Of the gene mutations studied, the KDR gene tended to be more common in malignant samples and may be a possible malignancy marker. Glass slides with FNAB samples were shown to be a reliable source of genetic material for NGS studies.
Introduction
Thyroid nodules may be detected in up to 67% of the adult population.1 Ultrasound examination remains the gold standard for a rapid diagnosis of potential malignancy.2,3 It may be complemented by fine‑needle aspiration biopsy (FNAB). Both methods enable a diagnosis and choice of appropriate treatment in 75% to 90% of patients.4 However, according to the Bethesda reporting system,5 cytologic results of FNAB are inconclusive in 10% to 25% of patients with thyroid nodules.6 These results include atypia or follicular lesion of undetermined significance (AUS/FLUS; Bethesda category III) and follicular neoplasm or suspicious for follicular neoplasm (FN/SFN; Bethesda category IV). In those cases, a differentiation between follicular thyroid adenoma and follicular thyroid cancer (FTC) is possible only on the basis of a histopathologic assessment of vascular or capsular invasion (or both),7 thus making surgery unavoidable.8
To avoid the need for a diagnostic surgery, new markers of presurgical differentiation between follicular thyroid adenoma and FTC are being extensively studied. Currently, one of the main areas for research is the identification of genetic alterations that may become markers of malignancy.9 The BRAF, 167 Gene Expression Classifier (GEC), and ThyroSeq tests are dedicated to the assessment of indeterminate thyroid nodules.10 However, they have not yet provided the results that would be sufficiently accurate for routine clinical use,10 while the comprehensive assessment of the genetic background of AUS/FLUS, FN/SFN, as well as FTC (not covered in the Cancer Genome Atlas Program) is still lacking.
The aim of the present study was to assess the genetic background of indeterminate thyroid nodules based on the material from FNAB samples in order to identify a mutational status that might facilitate the understanding of these nodules. We also aimed to discover novel genetic pathways potentially involved in FTC.
Patients and methods
Patient characteristics and clinicopathologic analysis
We retrospectively assessed 25 randomly selected white patients (2 men and 23 women; median age at diagnosis, 46 years [range, 28–68 years]) from the department of endocrinology at a single reference tertiary care hospital, who subsequently underwent thyroidectomy. The analysis covered the data collected between 2014 and 2018. We included patients who were preoperatively diagnosed with a thyroid nodule and who underwent FNAB, with a resulting cytologic diagnosis of an indeterminate thyroid nodule. Among the study group, 16 patients were diagnosed with FLUS, and 9 patients, with SFN, according to the most recent 2017 World Health Organization criteria.11
The study was approved by the Bioethical Committee of the Poznan University of Medical Sciences (decision no. 1016/15) and was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all patients.
The results of the cytologic analysis of FNAB specimens from the glass slides were paired with the results of the histopathologic assessment of samples obtained during total or subtotal thyroidectomy, and were subjected to further molecular analyses. The major exclusion criterion was incomplete medical records of a patient. Only patients who were not receiving any treatment at the time of diagnosis and who had no other endocrine disorders or cancer were included. The characteristics of patients are presented in Table 1.
Parameter | FLUS, Bethesda III (n = 16 [64%]) | SFN, Bethesda IV (n = 9 [36%]) | |
The differences between FLUS and SFN groups were nonsignificant.
Abbreviations: FLUS, follicular lesion of undetermined significance; SFN, suspicious for a follicular neoplasm | |||
Sex, male/female, n (%) | 2/14 (12/88) | 0/9 (0/100) | |
Age at diagnosis, y, median (range) | 45 (35–68) | 51 (28–65) | |
Final histopathologic diagnosis, n (%) | Benign | 13 (81) | 5 (56) |
Nodular hyperplasia | 10 (62) | 5 (56) | |
Follicular adenoma | 3 (19) | 0 | |
Malignant | 3 (19) | 4 (44) | |
Follicular thyroid cancer | 0 | 1 (11) | |
Papillary thyroid cancer | 3 (19) | 3 (33) | |
Malignancy rate, % | 18.8 | 44.4 | |
Length of follow‑up, mo, median (range) | 34 (13–57) | 30 (17–57) | |
Tumor size, mm, mean (SD) | 15.6 (9.3) | 19.8 (4.4) | |
Localization in the right/left lobe, n (%) | 8/8 (50/50) | 4/5 (44/66) | |
The diagnosis of FLUS and SFN was established on the basis of assessment by qualified pathologists and cytologic reevaluation of the FNAB specimens. For each patient, we recorded the age at diagnosis, sex, tumor size, multifocality, extrathyroidal extension, the presence of histopathologic signs of chronic lymphocytic thyroiditis (CLT), and histopathologic staging (pathological TNM) according to the 8th TNM classification.12 All data were analyzed according to final postoperative histopathologic diagnoses (Table 2).
Parameter | Benign outcome (n = 18 [72%]) | Malignant outcome (n = 7 [28%]) | P value | |
a Tumor stage according to the TNM classification
Abbreviations: NA, not applicable | ||||
Sex, male/female, n (%) | 2/16 (11/89) | 0/7 (0/100) | 0.48 | |
Age at diagnosis, y, median (range) | 46 (35–68) | 42 (28–65) | 0.64 | |
Age group, n (%) | <55 y | 13 (72) | 6 (86) | 0.64 |
≥55 y | 5 (28) | 1 (14) | ||
Follow‑up duration, mo, median (range) | 34 (14–57) | 23 (13–54) | 0.12 | |
Tumor size, mm, mean (SD) | 19.2 (8.8) | 12.6 (4.8) | 0.11 | |
Localization in the right lobe/left lobe, n (%) | 6/12 (33/67) | 6/1 (86/14) | 0.03 | |
Multifocality, n (%) | 4 (22) | 2 (29) | 1.00 | |
pT1aN0M0a, n (%) | – | 4 (57) | NA | |
pT2N0M0a, n (%) | – | 3 (43) | NA | |
Vascular invasion, n (%) | – | 5 (71) | NA | |
Chronic lymphocytic thyroiditis, n (%) | 6 (33) | 6 (86) | 0.03 | |
Mutations per patient, n, mean (SD) | 5.3 (2.0) | 4 (3.1) | 0.74 | |
Total number of genes with found mutations, n | 17 | 11 | 0.02 | |
Tumors were regarded as multifocal when 2 or more foci were found. In the case of multifocality, the size of the tumor was recorded as the size of the largest focus. All samples were anonymized prior to analysis.
Genomic DNA extraction
The areas of interest were indicated by a qualified pathologist from the 25 FNAB specimens; waste material after establishing a cytologic diagnosis, fixed with CytofixTM (BD Biosciences, San Jose, California, United States), and the unstained slides were manually microdissected. Genomic DNA was extracted using a QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, California, United States), according to the manufacturer’s instructions. Genomic DNA was quantified using a Qubit fluorometer (Invitrogen, Carlsbad, California, United States), and Nano Drop spectrometer (Thermo Fisher Scientific, Carlsbad, California, United States). The DNA quality, purity, and integrity were tested.
Next‑generation sequencing
The 50‑gene Ion AmpliSeq Cancer Hotspot Panel v2 (CHPv2; Thermo Fisher Scientific) with the Ion TorrentTM Personal Genome Machine platform (Thermo Fisher Scientific) was used for all experiments. The analytically and clinically validated panel enables to amplify 207 amplicons, covering approximately 2800 mutations deposited in the Catalogue of Somatic Mutations in Cancer (COSMIC) database from 50 oncogenes and tumor‑suppressor genes commonly mutated in human cancers listed in Supplementary material, Table S1. The Ion AmpliSeq Library Kit, v. 2.0 (Thermo Fisher Scientific), was used to amplify 10 ng of DNA according to the manufacturer’s instructions. Sequencing beads were templated and enriched using the Hi‑Q Template OT2 200 Kit (Thermo Fisher Scientific). The libraries were barcoded with the Ion Xpress Barcode Adaptors Kit (Thermo Fisher Scientific), clonally amplified by emulsion polymerase chain reaction in vitro on the Ion PGM Template OneTouch 2 system (Thermo Fisher Scientific). Ion Sphere Particles with DNA were isolated and sequenced on Ion 318v2 Chip using the Hi‑Q Sequencing Kit according to the manufacturer’s protocols (Thermo Fisher Scientific).
Mutation analysis
Data obtained from genomic experiments were subjected to analysis using dedicated software (Ion Torrent, Thermo Fisher Scientific).
Torrent Suite Software v.5.2 (Thermo Fisher Scientific) was used for signal processing, mapping, and quality control. The sequence variants were called and data were analyzed using the Ion Reporter with the AmpliSeq CHPv2 single‑sample workflow and default settings. The Variant Caller plugin included in Torrent Suite Software v.3.6 (Thermo Fisher Scientific), as well as the MutationTaster2 algorithm, were used to identify variations in target regions (http://www.mutationtaster.org). We categorized variants according to whether they comprised a stop codon, nonsynonymous, or frameshift mutation in the exonic region. Each of the identified genetic variations was coded according to the plus strand of the human genome assembly hg19 (Ensembl, www.ensembl.org). The limit of detection was a 5% mutational allelic frequency at 250 × coverage depth for each tested region.
To analyze a putative function of mutations as driver mutations, we used 4 separate programs: SIFT (Sorting Tolerant From Intolerant) algorithm,13 PolyPhen‑2,14 MutationTaster2,15 as well as FATHMM (Functional Analysis through Hidden Markov Models v2.3), which result in an index, calculated with a high‑throughput webserver, able to predict the functional consequences of coding variants (ie, nonsynonymous single nucleotide variants) and noncoding variants to distinguish between cancer‑promoting or driver mutations and other germline polymorphisms.
The following databases were for the presence of particular mutations and their previous reports: COSMIC v89, dbSNP v151, 1000 Genome Project, ClinVar, and ExAC v1.0.
Statistical analyses
The parameters were recorded and entered into a dedicated database. Descriptive analysis was used to summarize the collected data. To determine the normality of continuous variables, data were tested by the D’Agostino and Pearson omnibus normality test. We expressed the variables that were found to be normally distributed as means and respective standard deviations. Data that were found to be distributed otherwise were expressed as median and minimum–maximum values.
To compare differences between groups, the Fisher exact test (2 × 2 contingency table) for categorical variables was used. Interval data were compared using the Mann–Whitney test or the Kruskal–Wallis test with post‑hoc Dunn tests because the data did not follow a normal distribution. Odds ratios (ORs) and 95% CIs were calculated using the group of patients diagnosed with follicular adenomas as the reference population. We assessed a correlation between the number of mutations in a single patient and their age with the Pearson r correlation test and calculated the concordance rates.
A P value of less than 0.05 was considered significant. All statistical analyses were performed with StatSoft Statistica v12.0 (StatSoft, Kraków, Poland) and Analyse‑it for Microsoft Excel v3.53 software (Analyse‑it, Leeds, United Kingdom).
Results
The most common final benign diagnosis on histopathologic examination was a hyperplastic nodule (n = 15; 60%), followed by a follicular adenoma (n = 3; 12%). The most common malignant diagnosis was papillary thyroid cancer, diagnosed in 6 patients (24%): TNM stage pT1aN0M0 in 3 patients (50%) and pT2N0M0 in the other 3 patients (50%). The malignancy rate was 28% (18.8% and 44.4% for FLUS and SFN, respectively). The final malignant diagnosis more often coexisted with CLT than benign diagnosis (OR, 12; 95% CI, 1.16–123.69; P = 0.03; Table 2).
Any mutation from the 50‑gene Cancer Hotspot Panel v2 was identified in 14 patients, which constituted 56% of all FLUS and SFN samples. The RET mutation was detected in 11 patients (79% of the samples, regardless of the final outcome). The results showed that 10 samples (56% of all samples with benign diagnosis) harbored mutated genes. The most common was RET (n = 8; 80%), followed by TP53 (n = 7; 70%), FLT3 (n = 6; 60%), APC (n = 6; 60%), and PDGFRA (n = 5; 50%). In contrast, among the samples with malignant diagnosis, mutations were detected in 4 (24%). The most frequent were KDR and RET (both n = 3; 75%), followed by TP53 (n = 2; 50%). The KDR mutation tended to be more common in cancer samples than in benign ones (n = 3; 75% vs n = 2; 20%; P = 0.1). The ERBB4, IDH1, JAK3, PTEN, RB1, and SMAD4 mutations were only found in benign samples. There were no mutations found exclusively in cancer samples. The genetic landscape of both groups is presented in Figure 1.
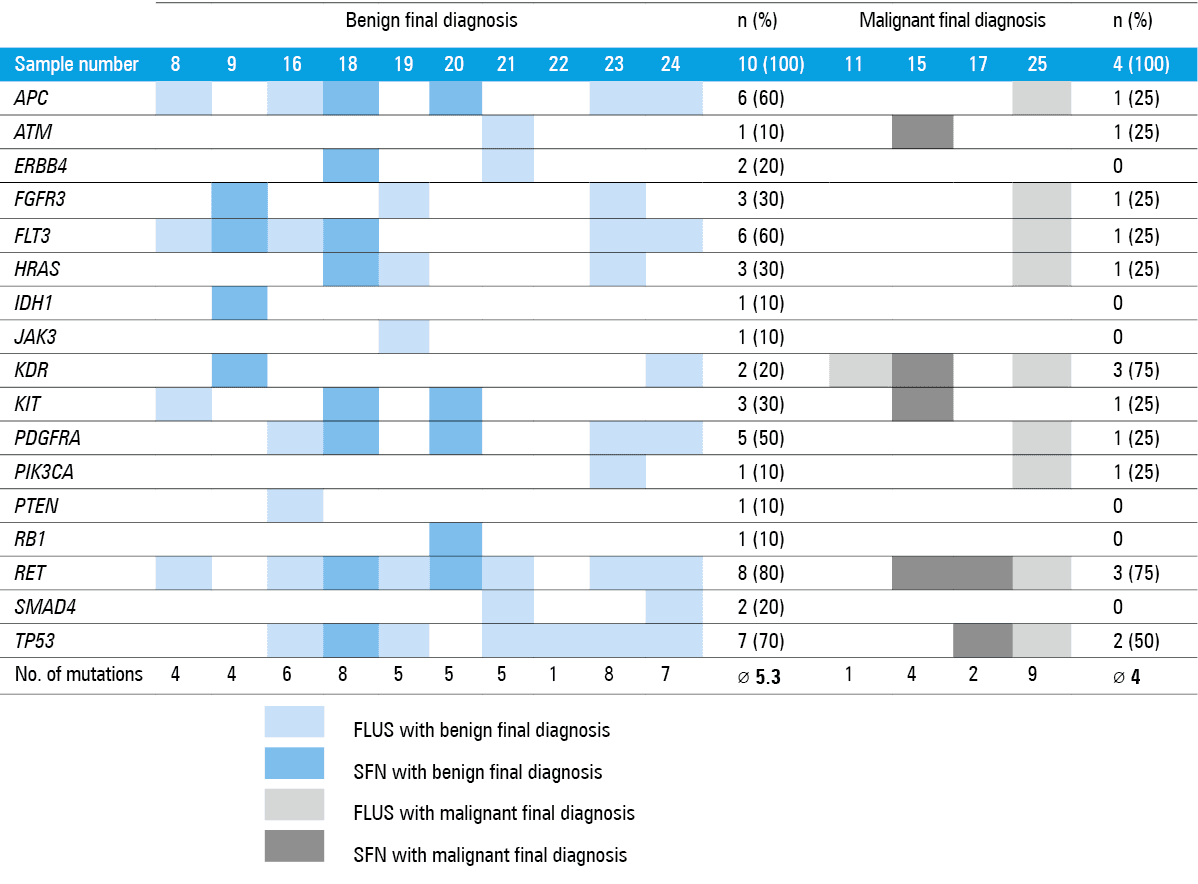
The total number of mutated genes was significantly higher in patients with benign tumors (17 vs 11, P = 0.02). The mean total number of mutations per every specimen did not differ between groups (4.9 mutations per sample for the entire group; range, 1–9). The distribution of mutations, type of genetic changes (point mutations [single nucleotide variants: intron, missense, synonymous variants] as well as duplications, insertions, and deletions), and the results of in silico analyses are presented in Table 3.
Gene | AAE | Type | Alteration (physical location) | DNA changes | Reference SNP | COSMIC mutation ID | Clinical significance | Previously reported | Benign final diagnosis (n = 10), n (%) | Malignant final diagnosis (n = 4), n (%) |
Abbreviations: AAE, amino acid exchange; ID, identifier; NA, not applicable; SNP, single nucleotide polymorphism; SNV, single nucleotide variant | ||||||||||
APC | p.G1494L | Insertion and deletion: missense variant | Chr5: 112175770 | c.4479_4480delGGinsAA; g.152553_152554delGGinsAA | rs1554085913 | – | Uncertain significance | Familial adenomatous polyposis 1 | 6 (60) | 1 (25) |
ATM | p.P858L | SNV: missense variant | Chr11: 108138003 | c.2572T>C; g.49445T>C | rs1800056 | COSM21826 | Benign/likely benign/ uncertain significance | Ataxia‑telangiectasia syndrome
Hereditary cancer‑predisposing syndrome | 1 (10) | 1 (25) |
ERBB4 | NA | SNV: intron variant | Chr2: 212812097 | g.212812097T>C | rs839541 | – | Not provided | Not specified | 2 (20) | 0 |
FGFR3 | p.T651T | SNV: synonymous variant | Chr4: 1807894 | c.1953A>G; g.17856A>G | rs7688609 | – | Benign | Not specified | 3 (30) | 1 (25) |
FLT3 | NA | SNV: intron variant | Chr13: 28602292 | g.28602292T>C | rs75580865 | – | Not provided | Not specified | 6 (60) | 1 (25) |
NA | SNV: intron variant | Chr13: 28610183 | g.28610183A>G | rs2491231 | – | Not provided | Not specified | |||
HRAS | p.H27H | SNV: synonymous variant | Chr11: 534242 | c.81T>C; g.6309T>C | rs12628 | COSM249860 | Benign | Costello syndrome
RASopathy | 3 (30) | 1 (25) |
IDH1 | p.G105G | SNV: synonymous variant | Chr2: 209113192 | c.315C>T; g.22607C>T | rs11554137 | NOCOSMIC105 | Benign | Not specified | 1 (10) | 0 |
JAK3 | p.V722I | SNV: missense variant | Chr19: 17945696 | c.2164G>A; g.18105G>A | rs3213409 | COSM34213 | Benign/likely benign/ likely pathogenic | Lymphoblastic leukemia, acute, with lymphomatous features
Severe combined immunodeficiency, autosomal recessive, T cell‑negative, B cell‑positive, NK cell‑negative
Acute megakaryoblastic leukemia | 1 (10) | 0 |
KDR | p.G472H | SNV: missense variant | Chr4: 55972974 | c.1416A>T; g.23789A>T | rs1870377 | – | Not provided | Not specified | 2 (20) | 3 (75) |
A | Insertion and deletion: intron variant | Chr4: 55962546 | g.55962547dup | rs3214870 | – | Not provided | Not specified | |||
KIT | p.M541L | SNV: missense variant | Chr4: 55593464 | c.1621A>C; g.74304A>C | rs3822214 | COSM28026
KIT COSM21983 | Benign/likely benign | Gastrointestinal stroma tumor
Chronic myelogenous leukemia
Mastocytosis
Partial albinism | 3 (30) | 1 (25) |
p.L546L | SNV: synonymous variant | Chr4: 55593481 | c.1638A>G; g.74321A>G | rs55986963 | COSM21983 | Benign/likely benign | Gastrointestinal stroma tumor
Mastocytosis
Partial albinism | |||
PDGFRA | p.P567P | SNV: synonymous variant | Chr4: 55141055 | c.1701A>G; g.50792A>G | rs1873778 | – | Benign | Gastrointestinal stroma tumor
Idiopathic hypereosinophilic syndrome | 5 (50) | 1 (25) |
p.V824V | SNV: synonymous variant | Chr4: 55152040 | c.2472C>T; g.61777C>T | rs2228230 | COSM22413 | Benign | Gastrointestinal stroma tumor
Idiopathic hypereosinophilic syndrome | |||
PI3CA | NA | SNV: intron variant | Chr3: 178917005 | g.178917005A>G | rs3729674 | – | Not provided | Not specified | 1 (10) | 1 (25) |
p.T1025T | SNV: synonymous variant | Chr3: 178952020 | c.3075C>T; g.90710C>T | rs17849079 | COSM21451 | Benign/likely benign | Cowden syndrome | |||
PTEN | p.A335T | SNV: nonsense variant | Chr10: 89720852 | c.1003C>T; g.102657C>T | rs121909231 | COSM5151 | Pathogenic | PTEN hamartoma tumor syndrome | 1 (10) | 0 |
RB1 | p.L569L | SNV: synonymous variant | Chr13: 49027140 | c.1707A>G; g.154258A>G | rs3092895 | – | Benign/likely benign/uncertain significance | Retinoblastoma
Hereditary cancer–predisposing syndrome | 1 (10) | 0 |
RET | p.L769L | SNV: synonymous variant | Chr10: 43613843 | c.2307G=; g.46327T>G | rs1800861 | – | Benign | Multiple endocrine neoplasia, type 2 | 8 (80) | 3 (75) |
p.S904S | SNV: synonymous variant | Chr10: 43615633 | c.2712C>G; g.48117C>G | rs1800863 | – | Benign | Not specified | |||
SMAD4 | NA | SNV: intron variant | Chr18: 48586344 | g.48586344C>T | rs948588 | – | Not provided | Not specified | 2 (20) | 0 |
TP53 | p.P72A | SNV: missense variant | Chr17: 7579472 | c.215C>G; g.16397C>G | rs1042522 | – | Drug response | Li–Fraumeni syndrome 1
Li–Fraumeni syndrome
Hereditary cancer‑predisposing syndrome | 7 (70) | 2 (50) |
Discussion
In our study, we assessed the genetic alterations of indeterminate thyroid nodules in preoperative FNAB specimens, using a large NGS panel. The results showed that the genetic heterogeneity of the FLUS and SFN samples in our patients corresponded with the heterogeneity of the final histopathologic diagnoses. A total of 17 mutated genes were found. Also, the number of mutations in individual samples was high, with a mean of 4.9 mutations per sample. This heterogeneity may be due to different routes of proliferation and differentiation,16 driven by completely different signals for benign and malignant diagnoses,17,18 as well as tumor heterogeneity.19 The latter can be divided into intertumor heterogeneity, which shows diverse genetic alterations based on the tumor sites, and intratumor heterogeneity, which contains different genetic alterations within the same tumor.20 Intratumor heterogeneity may derive from specific morphohistologic features, whereas at the molecular level, clonal intratumor heterogeneity originates from genomic instability, and nonclonal, from a microenvironment interaction.21 An extremely diverse genetic background, aggressive clone, and tumor heterogeneity in thyroid cancer have been described before.22-24
The overall malignancy rate in our patients was 28%. The previously described rates showed a considerable variation. For AUS/FLUS, the rates of 12%,25 26.6%,26 and 29.9%27 were reported, and for FN/SFN, 10.6%,28 20.8%,29 32.6%,30 37%,31 49%,32 and up to 53%.33 The discrepancies may relate to random variation, prior patient selection such as through referral bias, or institutional differences in pathologic interpretation.26 Nonetheless, the CIs reported in those series largely overlap with those in the current study, further supporting cancer incidence in FLUS and SFN shown by our group.
In recent years, significant advances have been made in thyroid cancer research. Molecular diagnostics of thyroid nodules extend the initial diagnostics of thyroid cancer, and molecular tests help distinguish between benign and malignant nodules, particularly in indeterminate FNAB results (Bethesda III–V).2 The fast‑evolving NGS technology offers a cost‑effective approach in cancer genomics and thyroid cancer.20 The NGS has already been used to study indeterminate thyroid nodules also based on DNA acquired from preoperative FNAB specimens.27,34,35 However, instead of commercially available thyroid‑specific panels such as the 7‑gene panel test, which covers a limited number of gene alterations (BRAF and NRAS/HRAS/KRAS point mutations and RET/PTC and PAX8/PPARg translocations),36 or its successors (v2, v2.1, and v3),37 we used a large NGS panel of not only thyroid‑specific genes. The panel, in addition to BRAF, RAS, and RET hotspot mutations, covers numerous other genes involved in thyroid carcinogenesis and malignant progression (eg, AKT1, APC, ATM, CTNNB1, PI3CKA, PTEN, RB1, and TP53).38-40 The quality of DNA obtained from cytology specimens is considered to be better than that of formalin‑fixed paraffin‑embedded tissue.20 Besides, as previously reported, NGS can also provide information on routine smears that have suboptimal DNA input quantity and quality as well as post‑sequencing metrics.41
Ultrasound features considered to be predictors of malignancy do not allow a reliable differentiation between malignant and benign nodules,3 neither do the GEC or the ThyroSeq v2 tests.42 Therefore, our goal was to identify novel genetic pathways potentially involved in FTC. KDR and RET (both present in 75% of samples), followed by TP53, were among the most common mutated genes found in our study in patients with a cancer diagnosis. KDR tended to be more common in malignant than benign samples, and as such, it appears to be a possible candidate as a genetic marker for malignancy. Its role was previously reported in breast cancer.43 KDR encodes kinase insert domain receptor of the vascular endothelial growth factor, which acts as a mediator in the regulation of angiogenesis, vascular development, vascular permeability, and embryonic hematopoiesis, as well as promotes proliferation, survival, migration, and differentiation of endothelial cells. KDR gene mutations may play a major role in tumor angiogenesis.44
The number of mutated genes was higher in the group with benign diagnosis. This may indicate the complex genetic background not only of thyroid cancers but also of follicular adenomas or other benign nodules. For instance, a determination of the tumors and malignant transformation is still related to common KDR, RET, and TP53 alterations, which may result from subclonal evolution. Both KDR and RET encode kinase domain receptors. The important presence of the RET gene mutation and its possible diagnostic value for identifying benign tumors have been previously described by our team.45 However, the high level of genomic dysregulation found in benign tissue, as reflected by a high number of identified mutations and exclusive contribution of the mutated ERBB4, IDH1, JAK3, PTEN, RB1, or SMAD4 genes (not found in malignant cases) as the markers of indolent thyroid tumors, requires further comprehensive studies. The lack of the BRAF mutation, reported also in other FTC studies,46 and the occurrence of KDR and RET may encourage further research of new genetic pathways of follicular malignancy. The in silico analyses showed that numerous mutations commonly occur in other malignancies, showing a possible intersection of cancer pathways, beyond the previously known ones for thyroid cancer. The detailed description of genetic findings shows that the mutations interpreted by various algorithms as benign or possibly benign have been reported to occur in various other malignancies, which may prove that the role of particular mutations may be more complex and our understanding of them still incomplete. The specific role of each variant is to be determined in functional studies.
Another interesting finding is that the malignant final diagnosis showed a high degree of correlation with CLT. This is in line with previous reports showing that in patients with differentiated thyroid cancer, the occurrence of CLT was higher than in those with benign thyroid diseases.47 This link was also studied in detail regarding CLT‑associated clinicopathologic features of thyroid cancer.48
Although the Polish population is in part considered to have iodine depletion,49 previous research showed no differences in genetic alterations of thyroid cancers between iodine‑rich and iodine‑deficient countries, which may suggest that iodine intake might not affect the genetic alterations of differentiated thyroid cancer.50 This makes our study findings even more generalizable.
The major limitation of our study is the small number of the analyzed cases. Therefore, the conclusions should be formulated with caution, and a much larger group of patients is needed to clarify the clinical importance of the findings. However, this preliminary study emphasizes the potential utility of applying a broader NGS panel to describe the genetic background of indeterminate thyroid nodules. Despite the lower statistical power, the present results indicate the genetic heterogeneity of indeterminate thyroid nodules. Aiming to increase the homogeneity of the group and the reliability of results, we opted only for the FLUS and SFN samples.
In conclusion, our study shows that the heterogeneity of the genetic alterations of indeterminate thyroid nodules corresponds to its histopathologic heterogeneity, which may be even higher for benign tumors. The role of KDR as a potential malignancy marker needs to be confirmed, as does the possible role of ERBB4, IDH1, JAK3, PTEN, RB1, or SMAD4 as the markers of indolent thyroid tumors. Glass slides with FNAB samples may constitute a reliable source of genetic material for NGS analysis. The new genetic pathways for FTC should be searched for using new sequencing technologies, such as NGS with large gene panels, to improve our understanding of the genetics of thyroid nodules and its overlap with other malignancies. Consequently, it may facilitate a more effective preoperative cancer diagnosis of thyroid nodules.
- Tamhane S, Gharib H. Thyroid nodule update on diagnosis and management. Clin Diabetes Endocrinol. 2016; 2: 17. | Crossref
- Jarząb B, Dedecjus M, Słowińska‑Klencka D, et al. Guidelines of Polish national societies Diagnostics and Treatment of Thyroid Carcinoma. 2018 update. Endokrynol Pol. 2018; 69: 34‑74.
- Woliński K, Szkudlarek M, Szczepanek‑Parulska E, Ruchała M. Usefulness of different ultrasound features of malignancy in predicting the type of thyroid lesions: a meta‑analysis of prospective studies. Pol Arch Med Wewn. 2014; 124: 97‑104.
- Bongiovanni M, Spitale A, Faquin WC, et al. The Bethesda system for reporting thyroid cytopathology: a meta‑analysis. Acta Cytol. 2012; 56: 333‑339. | Crossref
- Cibas ES, Ali SZ. The 2017 Bethesda system for reporting thyroid cytopathology. Thyroid. 2017; 27: 1341‑1346. | Crossref
SUPPLEMENTARY MATERIAL
ARTICLE INFORMATION