When rare matters: aortic valve destruction in myeloperoxidase antineutrophil cytoplasmic antibody–associated microscopic polyangiitis

 CC BY 4.0
CC BY 4.0
When rare matters: aortic valve destruction in myeloperoxidase antineutrophil cytoplasmic antibody–associated microscopic polyangiitis
Microscopic polyangiitis (MPA) is an antineutrophil cytoplasmic antibody (ANCA)-associated, pauci‑immune necrotizing small vessel vasculitis with multisystem involvement, most commonly renal, pulmonary, and cutaneous. Its clinical presentation is heterogeneous and often nonspecific, with constitutional symptoms, such as fever, unintentional weight loss, and asthenia.1
We report a case of a 55‑year‑old woman with microscopic polyangiitis, diagnosed in October 2024, with the presence of strongly positive antimyeloperoxidase antibodies (MPO‑ANCA).
At the time of diagnosis, the patient presented with pulmonary symptoms (hemoptysis, cough) and microscopic hematuria, as well as a history of myocarditis diagnosed in July 2024, confirmed on cardiac magnetic resonance imaging (MRI) showing subepicardial and mid‑wall late gadolinium enhancement (LGE) consistent with nonischemic myocarditis. This finding suggested inflammatory myocardial involvement, although without evidence of active inflammation. Notably, cardiac MRI can detect subclinical myocardial injury in ANCA‑associated vasculitis, yet the clinical significance of LGE remains uncertain, as enhancement may persist or progress despite apparent clinical remission, and it is not necessarily linked to adverse outcomes.2 High‑resolution computed tomography (CT) of the chest performed in July 2024 demonstrated several small ground‑glass nodules in both lungs, exhibiting a peribronchovascular distribution and a tendency toward consolidation, which indicated pulmonary involvement. Renal involvement was also present, with an active urinary sediment and serum creatinine concentrations between 0.8 and 1.2 mg/dl (reference range [RR], 0.5–1.1 mg/dl). Serum MPO‑ANCA level was above 168 IU/ml (RR <10 U/ml). Based on the 2022 American College of Rheumatology / European Alliance of Associations for Rheumatology (ACR/EULAR) classification criteria,3 a diagnosis of MPA was established.The patient scored 9 points: 6 points for serum MPO‑ANCA and 3 points for the presence of interstitial lung disease. MPO‑ANCA positivity is not specific to MPA and can also be present in eosinophilic granulomatosis with polyangiitis (EGPA). However, EGPA was excluded based on the absence of asthma, lack of peripheral eosinophilia, and no evidence of nasal polyps on otolaryngological examination and CT of the paranasal sinuses. The absolute eosinophil count at the time of MPA diagnosis was 120/µl (RR, 30–370/µl), well below the 1000/µl threshold required by the 2022 ACR/EULAR classification criteria for EGPA.
Therapy was initiated in October 2024. It involved intravenous methylprednisolone pulses, oral prednisone, and intravenous cyclophosphamide, according to the European Vasculitis Study Group protocol,4 followed by azathioprine for maintenance. This approach aligns with contemporary standards of treatment.
In May 2025, the patient was readmitted to a hospital with exertional intolerance corresponding to New York Heart Association (NYHA) class II–III. Transthoracic echocardiography demonstrated severe aortic valve regurgitation (Figure 1A and 1B). Blood cultures remained negative; however, infective endocarditis (IE) could not be definitively excluded. Therefore, transesophageal echocardiography was performed, showing destruction of the aortic valve, with no evidence of valvular vegetations. No clinical activity of vasculitis was detected. After clinical stabilization, the patient was discharged home.
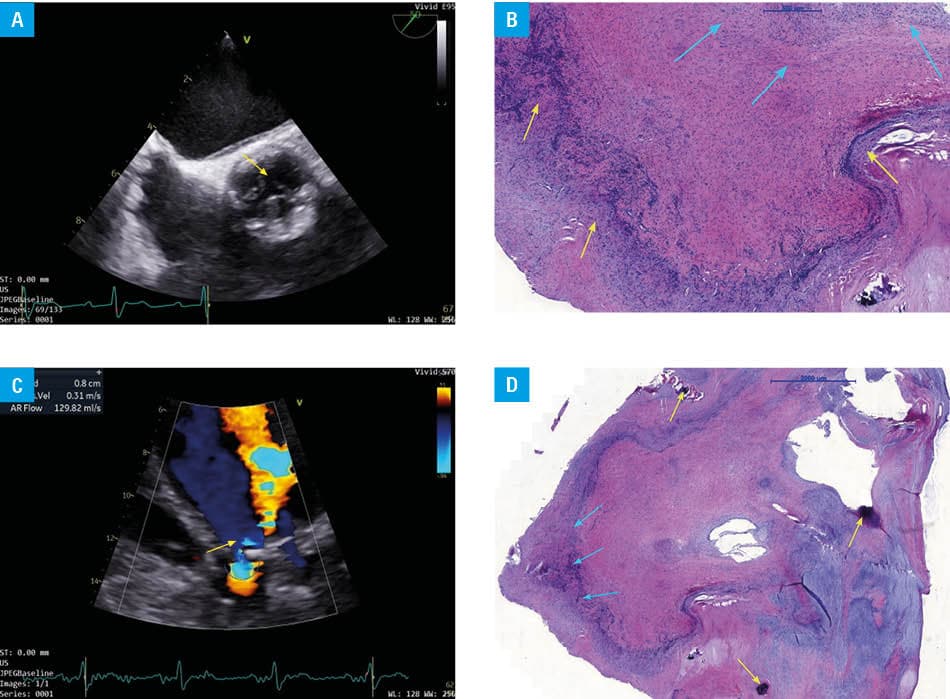
In July 2025, the patient presented again with acute nocturnal dyspnea and symptoms indicating progression of heart failure (NYHA class III–IV). Despite optimized, guideline‑directed medical therapy for heart failure, her clinical condition deteriorated. The decision of surgical intervention was made, and surgical aortic valve replacement was performed on August 17, 2025. The native aortic valve was submitted for histopathological examination (Figure 1C and 1D), which demonstrated an inflammatory process involving the cups of the aortic valve.
Cardiac involvement in MPA is uncommon and can manifest as pericarditis, myocarditis, or isolated valvular disease. In published series and case reports, endocardial involvement in MPA most commonly affects the aortic valve, followed by the mitral valve.4 Although cardiac involvement in MPA is rare and largely limited to case reports, vasculitic etiologies should be carefully considered when evaluating deteriorating cardiac function in affected patients.
- Kidney Disease: Improving Global Outcomes (KDIGO) ANCA Vasculitis Work Group. KDIGO 2024 Clinical Practice Guideline for the Management of Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis. Kidney Int. 2024; 106: 160‑163. | Crossref
- Karageorgiou I, Bhatia U, Alakhras H, et al. Cardiac magnetic resonance imaging findings in patients with antineutrophil cytoplasmic antibody‑associated vasculitides: a systematic review. ACR Open Rheumatol. 2025; 7: e70026. | Crossref
- Suppiah R, Robson JC, Grayson PC, et al. DCVAS INVESTIGATORS. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for microscopic polyangiitis. Ann Rheum Dis. 2022; 81: 321‑326. | Crossref
- Jeantin L, Lenfant T, Bataille P, et al; French Vasculitis Study Group. Antineutrophil cytoplasm antibody‑associated vasculitides valvular impairment: multicenter retrospective study and systematic review of the literature. J Rheumatol. 2022; 49: 1349‑1355. | Crossref
ARTICLE INFORMATION