Jeśli tylko okaże się, że doświadczenie stanu Waszyngton zaowocuje wysoką wykrywalnością choroby Wilsona, nieprzystającą do szacowanej epidemiologii, warto będzie pomyśleć o realizacji podobnego programu przesiewowego w Polsce – mówi prof. Adam Przybyłkowski, kierownik Kliniki Gastroenterologii i Chorób Wewnętrznych WUM.

Prof. Adam Przybyłkowski. Fot. Michał Teperek / WUM
Jerzy Dziekoński: Jakich trudności diagnostycznych przysparza lekarzom choroba Wilsona w przypadku pacjentów dorosłych i jak często, w związku z tym, dochodzi do fałszywych rozpoznań? Jak długo może trwać opóźnienie diagnostyczne?
Prof. Adam Przybyłkowski: Znam tylko jednego pacjenta, któremu fałszywie rozpoznano chorobę Wilsona. Natomiast w większości przypadków rozpoznania nie postawiono przez lata mimo ewidentnych objawów tej choroby. Lekarze wszystkich specjalności mają skądinąd pragmatyczny zwyczaj ignorowania nieznacznie podwyższonej aktywności aminotransferaz. Wynika to po części z wiedzy wyniesionej ze studiów, ale też np. definicji uszkodzenia wątroby używanego między innymi w badaniach klinicznych, gdzie dopiero kilkukrotne zwiększenie aktywności aminotransferaz jest objawem alarmującym. Stąd bardzo często widzimy pacjentów pediatrycznych, którzy czekali 3-4 lata na diagnozę, bo podwyższone wyniki bilirubiny i aminotransferaz, bez zagłębiania się w temat, tłumaczono zaburzeniami wieku rozwojowego. U dorosłych opóźnienia w rozpoznaniu choroby są wieloletnie, podczas gdy przy pomocy bardzo prostych i niedrogich badań, czyli oznaczenia stężenia ceruloplazminy czy dobowego wydalania miedzi z moczem, można bardzo szybko dotrzeć do sedna problemu, czyli do diagnozy choroby Wilsona.
Jak rozumiem, w przypadku pacjentów pediatrycznych ma to o tyle znaczenie, że wcześniejsze zaopiekowanie się chorobą oznacza brak objawów neurologicznych czy też psychiatrycznych w późniejszym, dorosłym życiu.
W leczeniu tej choroby współpracujemy z lekarzami z II Kliniki Neurologicznej Instytutu Psychiatrii i Neurologii, z zespołem kierowanym przez prof. Annę Członkowską oraz z zespołem pediatrów pod kierownictwem prof. Piotra Sochy z Centrum Zdrowia Dziecka. Wychodzimy z założenia, że rozpoznana we wczesnym wieku i prawidłowo leczona choroba nie da żadnych objawów. A jeśli wystąpią, to w minimalnym stopniu. Dlatego tak bardzo zależy nam na tym, żeby chorobę Wilsona diagnozować w fazie presymptomatycznej. Staramy się, żeby w jak najmniejszym stopniu ingerować w życie pacjentów. Zgodnie z modelowym schematem opieki, kontrole odbywają się raz do roku. Bo jeśli wszystko jest w porządku, nie ma potrzeby częstszego wykonywania badań.
W jaki zatem sposób dotrzeć do pacjentów na etapie, kiedy choroba Wilsona nie niesie jeszcze żadnych poważnych powikłań?
W stanie Waszyngton w Stanach Zjednoczonych w pierwszym miejscu na świecie już w przyszłym roku wystartuje program badań przesiewowych noworodków. Wykorzystany zostanie test z suchej kropli krwi, który pozwoli, podobnie jak u nas w przypadku fenyloketonurii, na wyłowienie chorych na chorobę Wilsona. Wstępne wyniki metody opartej o wykrywanie fragmentu białka ATP7B są bardzo obiecujące. Czułość i specyficzność tego testu przekracza 90%.
Czy są miejsca na ziemi – jak np. stan Waszyngton – gdzie choroba Wilsona występuje częściej? Jak przedstawia się jej epidemiologia w Polsce?
Schemat dziedziczenia choroby Wilsona jest dosyć prosty, natomiast powstawanie nowych mutacji i ich geograficzne zróżnicowanie stanowi dla genetyków i ewolucjonistów pewną zagadkę. Przyjmuję się, że częstość występowania tej choroby to 1 chora/chory na 30 tys. osób. Jak łatwo policzyć, w Polsce powinniśmy mieć pod opieką ok. 1,2 tys. osób z chorobą Wilsona. Przez lata chorzy byli skoncentrowani właściwie w jednym ośrodku, czyli we wspomnianej II Klinice Neurologicznej IPiN. Prof. Ignacy Wald, który niegdyś tam pracował, odkąd dowiedział się w latach 50-tych XX wieku o istnieniu tej choroby, jeździł po klinikach neurologicznych i psychiatrycznych w kraju i aktywnie wyszukiwał pacjentów. Później przez ostatnich 50 lat opiekę na chorymi przejęła i rozwinęła prof. Anna Członkowska. W wyszukiwanie pacjentów, po wykładzie prof. Członkowskiej na temat występowania choroby Wilsona u dzieci, włączyli się również pediatrzy z Centrum Zdrowia Dziecka. I to są dwa główne ośrodki w Polsce zajmujące się diagnozowaniem i leczeniem choroby Wilsona. Ośrodek, w którym pracuję, dołączył jako trzeci. W wymienionych 3 placówkach leczonych jest ok. 600 pacjentów, czyli co najmniej połowa chorych pozostaje nierozpoznana. Dane z populacji, w których na szeroką skalę wykonuje się badania genetyczne, jak np. populacja estońska, potwierdzają, że wiele osób może pozostawać bez diagnozy.
Jeśli chodzi o obszary na świecie, gdzie częstość występowania choroby Wilsona jest znacznie większa, zazwyczaj są to tereny geograficznie odizolowane. Takim przykładem jest Sardynia albo wieś na wyspie Gran Canaria, która od pozostałej części lądu jest oddzielona wąwozem. Przeszkody geograficzne sprawiły, że tradycyjnie zawierano tam związki w dość bliskim stopniu pokrewieństwa. Kolejny przykład to Kostaryka. Poza tym jeszcze Korea i w ogóle Azja jest kontynentem o większej częstości występowania choroby Wilsona.
Czy, Pańskim zdaniem, wprowadzenie badań przesiewowych noworodków w Polsce miałoby sens?
Uważam, że tak. Skoro mamy szansę zidentyfikować chorobę Wilsona co najmniej u kilkuset osób, to dlaczego mielibyśmy tego nie zrobić. Mamy przecież doskonałe efekty leczenia. Trudno przejść obojętnie obok tak trudnych powikłań, skoro część z nich może być nieodwracalna. My, lekarze zajmujący się tą chorobą, proaktywnie namawiamy członków rodzin pacjentów, żeby pozwolili się zbadać w kierunku choroby Wilsona. Telefonujemy do nich, wysyłamy listy. Dosłownie w tym tygodniu przyjęliśmy pacjenta, który jest bratem innej pacjentki, i u którego rozpoznaliśmy chorobę Wilsona w wieku 41 lat. A rekordzista, u którego ją zdiagnozowaliśmy, miał ponad 70 lat. Pacjenci, którzy są diagnozowani po 50. roku życia, wcale nie należą do rzadkości.
Jak Pan wspomniał, leczenie jest na tyle skuteczne, że można zapobiec wystąpieniu objawów choroby. Jakie terapie są dzisiaj dostępne dla pacjentów w Polsce?
Leczenie jest proste i polega na przyjmowaniu leków doustnych, zwykle w podzielonej dawce dobowej. Do wyboru są trzy preparaty: D-penicylamina, która chelatuje miedź i powoduje jej usuwanie z organizmu; sole cynku, które zapobiegają wchłanianiu miedzi z przewodu pokarmowego i doprowadzają niejako do niedoboru tego pierwiastka oraz trientyna, lek w mechanizmie działania podobny do D-penicylaminy. W terapii kłopotliwe może być jedynie to, że trzeba zachowywać odstęp między posiłkiem a przyjmowanym lekiem – godzinę przed posiłkiem albo dwie godziny po posiłku. Pacjenci, którzy stosują się do naszych zaleceń, poza przyjmowaniem leków żyją zupełnie tak jak zdrowi ludzie. Różnicę stanowią właśnie te 4 tabletki dziennie.
W jaki sposób choroba rozwija się u osób, u których nie została rozpoznana i które nie przyjmują leków?
Dochodzi do uszkodzenia wątroby, które przybiera bardzo różne postacie: od przewlekłego zapalenia wątroby, czy tlącego się wzrostu aktywności enzymów wątrobowych po piorunującą niewydolność wątroby, którą leczy się bardzo trudno – zwykle konieczny jest przeszczep, aczkolwiek są doniesienia o leczeniu plazmaferezami.
Tlące się, nierozpoznane zapalenie wątroby doprowadza finalnie do marskości i do wystąpienia jej wszystkich objawów, czyli żylaków przełyku wraz z krwawieniem, wodobrzuszem, żółtaczką, zaburzeniami krzepnięcia i wszelkimi innymi zmianami wynikającymi z marskości.
Nieleczona choroba u połowy pacjentów, według niektórych szacunków nawet u 60%, doprowadza do wystąpienia objawów neurologicznych takich jak nadmierne ślinienie się, drżenia i przeróżnego rodzaju inne objawy ze strony układu pozapiramidowego, które, jeśli trwają długo, stają się nieodwracalne.
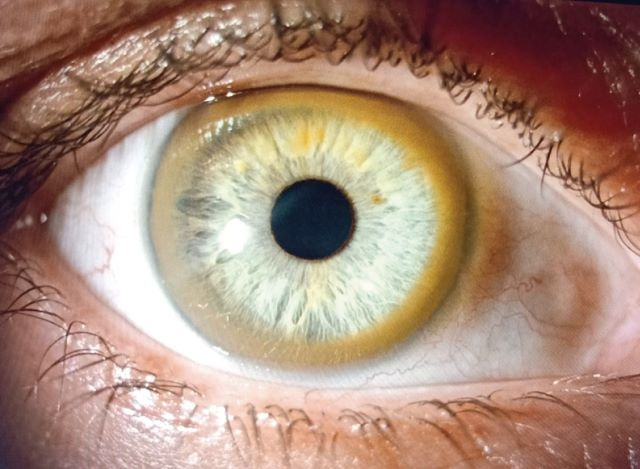
Charakterystycznym objawem choroby Wilsona jest tzw. pierścień Kaysera i Fleischera występujący praktycznie u 100% chorych z zajętym ośrodkowym układem nerwowym. U chorych na postać wątrobową występuje mniej więcej w połowie przypadków. W żaden sposób nie upośledza widzenia i zwykle wycofuje się w trakcie skutecznego leczenia przeciwmiedziowego.
Można zatem powiedzieć, że lekarze innych specjalności niż gastrolodzy czy neurolodzy mają rolę do odegrania w rozpoznawaniu choroby Wilsona.
Opiekujemy się ponad 300 pacjentami chorymi na chorobę Wilsona w naszej klinice i pośród nich jest jeden lekarz, któremu po 40. roku życia chorobę Wilsona rozpoznała koleżanka okulistka. Pacjent doskonale funkcjonował, nie miał żadnych objawów. I chociaż w badaniach krwi występowała nieprawidłowa aktywność aminotransferaz, nikt się tym nie zainteresował. Dzięki temu, że okulistka rozpoznała pierścień Kaysera-Fleischera, został zdiagnozowany i rozpoczęliśmy skuteczne leczenie.
Ta granica 40. rż. po raz kolejny pojawia się w naszej rozmowie. Dlaczego to takie istotne, żeby rozpoznanie było jednak przed jej przekroczeniem?
Choroba powinna być rozpoznana jak najwcześniej. Zwykle po 30. roku życia a prawie u wszystkich nieleczonych chorych po 40. r.ż. wątroba jest już przebudowana. Dochodzi do włóknienia, a u niektórych chorych obecna jest już ewidentna przebudowa marska. Takich chorych leczymy inaczej, bo nie tylko dbamy o prawidłową gospodarkę miedziową, ale również obserwujemy ich pod kątem marskości, rozwoju raka wątrobowo-komórkowego, czy nadciśnienia wrotnego. Tacy chorzy wymagają częstszego monitorowania aniżeli raz do roku. Modyfikacja stylu życia jest już bardziej inwazyjna.
Czy postać neurologiczna jest wtórna do postaci wątrobowej choroby?
Na ten temat toczą się bardzo ciekawe dyskusje. Jeśli założymy, że wątroba jest narządem magazynującym nadmiar miedzi, w przebiegu choroby Wilsona zaczyna chorować jako pierwsza. Osobiście zgadzam się z takim podejściem. Kiedy zdolności do magazynowania miedzi w wątrobie się wyczerpują, więcej miedzi niezwiązanej z ceruloplazminą, tzw. miedzi wolnej, zaczyna krążyć we krwi. I dopiero to doprowadza do uszkodzeń neurologicznych. Nikt tego nie udowodnił, ale jest taka sekwencja czasowa, że objawy neurologiczne zwykle pojawiają się u osób starszych, częściej u mężczyzn niż u kobiet. Te wszystkie dane pochodzą z czasów, kiedy diagnostyka była jeszcze nie tak czuła jak dziś. Jeszcze w latach 80-tych i 90-tych ubiegłego wieku znakomita większość chorych w Polsce prezentowała przy rozpoznaniu objawy neurologiczne. Wynikało to z tego, że objawy neurologiczne bardziej zmieniały życie pacjentów; były bardziej widoczne i alarmujące. Dlatego neurolodzy wcześniej rozpoznawali chorobę Wilsona niż gastroenterolodzy, czy lekarze innych specjalności. Zapalenie wątroby może tlić się latami w sposób praktycznie bezobjawowy. Wydaje mi się, że nie ma ani jednego pacjenta opisanego na świecie, ale mogę się mylić, który miałby tylko czystą postać neurologiczną bez zajęcia wątroby. Owszem, pojawiają się w literaturze przypadki bez zajęcia wątroby, ale wiązało się to przede wszystkim z tym, że diagnostyka uszkodzenia wątroby nie została przeprowadzona w oparciu o odpowiednio czułe narzędzia. Według aktualnych statystyk amerykańskich, przy rozpoznaniu około 70% chorych manifestuje objawy wątrobowe, a 52% objawy wątrobowe i neurologiczne. Jedynie jedna piąta chorych przy rozpoznaniu ma dominujące objawy neurologiczne i psychiatryczne. Według statystyk opublikowanych przez zespół Instytutu Psychiatrii i Neurologii, który zamieścił bardzo ciekawą pracę o siedmiu dekadach leczenia choroby Wilsona w Polsce, mniej więcej do końca lat 90. objawy neurologiczne jako przyczyna rozpoczęcia diagnostyki były opisywane u 53% chorych, a objawy wątrobowe – u 32%. Pacjenci bezobjawowi stanowili 14%. Natomiast począwszy od lat dwutysięcznych, objawy wątrobowe były obserwowane u 44%, a neurologiczne u 36% chorych. Zwiększenie czujności lekarzy i postęp diagnostyki laboratoryjnej powodują, że coraz częściej rozpoznajemy pacjentów bez objawów neurologicznych.
Czy choroba Wilsona manifestuje się też w innych układach poza wątrobą i ośrodkowym układem nerwowym?
To już ustaliliśmy, że w oku, w głowie i w wątrobie daje szerokie spektrum objawów. Mamy pod opieką chorych, którzy bez objawów neurologicznych manifestowali objawy psychiatryczne typu ostre psychozy, zaburzenia lękowe czy zaburzenia afektywne. Choroba Wilsona może manifestować się niedokrwistością hemolityczną, choć zwykle objawy te towarzyszą ostrej niewydolności wątroby. Zdarzają się również objawy kostno-stawowe, czyli artropatia. Może pojawić się niewydolność serca i zaburzenia rytmu serca, niedoczynność przytarczyc, tłuszczaki w tkance podskórnej. Może wystąpić kwasica nerkowa przyjmująca postać zespołu Fanconiego.
W zasadzie mamy cały pakiet.
Tak, mnogość tych objawów powoduje, że chorzy trafiają do nas z różnych stron i z różnych kierunków diagnostycznych. Dlatego uważam, że badania przesiewowe są bardzo potrzebne. Jeśli tylko okaże się, że doświadczenie stanu Waszyngton zaowocuje wysoką wykrywalnością choroby Wilsona, nieprzystającą do szacowanej epidemiologii, to bez dwóch zdań warto będzie pomyśleć o realizacji podobnego programu screeningowego w Polsce.
Na koniec pozostaje zapytać o to, o co powinienem był zapytać na początku, czyli o patomechanizm powstawania choroby Wilsona?
Patomechanizm jest fascynujący podobnie jak cała choroba Wilsona. Na skutek mutacji genu, który koduje białko ATP7B dochodzi do nieprawidłowego transportu miedzi w wielu różnych komórkach organizmu. Białko ATP7B jest kluczowym transporterem w hepatocycie. Jeśli białko ulega degradacji – ma nieprawidłową strukturę lub uszkodzone fragmenty wiążące ATP – nie transportuje miedzi do żółci. Miedź nie jest usuwana z hepatocytów i nie następuje wbudowywanie miedzi do ceruloplazminy. A to są dwie podstawowe funkcje tego białka ATP7B. Z powodu utraty pierwszej funkcji, w organizmie gromadzi się nadmiar tego pierwiastka, natomiast brak dostarczania jonów miedzi do apoceruloplazminy powoduje, że ceruloplazmina jest bardzo szybko degradowana i jej stężenie w surowicy się obniża. Kłopot z chorobą Wilsona w odróżnieniu od innych uwarunkowanych genetycznie, ale występujących częściej chorób, polega na tym, że mutacji w genie APT7B kodującym białko o tej samej nazwie opisano już ponad 1200. Nie wszystkie są patogenne, ale wcześniej, kiedy diagnostyka genetyczna była bardzo droga, diagnoza tej choroby na poziomie genetycznym była bardzo trudna. Dzisiaj dzięki sekwencjonowaniu całego genu badanie genetyczne stało się to znacznie bardziej dostępne cenowo i geograficznie. Niemniej jednak interpretacja wyników nadal jest trudna i zawsze powinna być korelowana z parametrami metabolizmu miedzi, czyli z oznaczaniem stężenia miedzi w surowicy. Idealnie byłoby gdybyśmy wykonywali oznaczenie miedzi niezwiązanej z ceruloplazminą. Na razie żaden ośrodek w Polsce takich badań nie wykonuje i dopiero przymierzamy się do uruchomienia tych oznaczeń w dwóch ośrodkach w Warszawie. No i zawsze warto wykonać dobowe wydalanie miedzi z moczem, które jest bardzo dobrym parametrem.
Jeszcze jednym badaniem, które jest równie czułe co dobowe wydalanie miedzi z moczem, jest test z miedzią radioaktywną, który polega na podaniu takiej miedzi pacjentowi i pomiarze radioaktywności krwi w odpowiednich odstępach czasowych. Z racji niskiej dostępności tego badania i słabego rozpowszechnienia na świecie, traci ono na znaczeniu.
Czy na horyzoncie pojawiają się nowe, obiecujące terapie, które mogłyby zrewolucjonizować leczenie choroby Wilsona?
Pacjenci z chorobą Wilsona w ogóle wydają się być obiecującą populacją, jeśli chodzi o wdrożenie terapii genetycznych. W dwóch amerykańskich ośrodkach prowadzone są badania nad różnymi wektorami wirusowymi transportującymi gen ATP7B w różny sposób. Wyniki są obiecujące. Wydaje się, że podobnie jak w niektórych rzadkich chorobach neurologicznych, gdzie terapia genowa jest już właściwie wdrożona i zarejestrowana przez Europejską Agencję Leków, pacjenci z chorobą Wilsona są na początku peletonu w wyścigu do skutecznego leczenia. Myślę, że niedługo doczekają się terapii, która poprzez cykliczne iniekcje ustabilizuje gospodarkę miedzi w organizmie.
Wchodzi również terapia oparta o technikę CRISPR, gdzie szansa na trwały efekt leczniczy, zwłaszcza u pacjentów, którzy są homozygotami, zdaje się być znacznie większa.
Warto też powiedzieć, że zanim doczekamy się nowych terapii, dostępne dzisiaj leki powinny być ordynowane przez lekarzy z doświadczonych ośrodków, ponieważ leczenie choroby Wilsona może być w dwóch sytuacjach niebezpieczne. Jeśli na początku wprowadzamy dużą dawkę leków chelatujących, może wystąpić nieodwracalne neurologiczne pogorszenie stanu pacjenta, wynikające najprawdopodobniej z dużej mobilizacji miedzi. Dlatego leczenie, jeśli tylko stan wątroby na to pozwala, należy wprowadzać powoli.
Z kolei, głównie w przypadku cynku, zwłaszcza w terapii prowadzonej przez wiele lat, występuje ryzyko doprowadzenia do jatrogennego niedoboru miedzi, który również może skutkować uszkodzeniem ośrodkowego układu nerwowego.
Trzeci bardzo ważny aspekt to współpraca między lekarzem a pacjentem. Ci chorzy często prezentują objawy zaburzeń osobowości, czasem są bardzo emocjonalni i trzeba do nich podchodzić bardzo cierpliwie. Przerwanie ciągłości terapii z reguły kończy się źle, również dramatycznymi pogorszeniami neurologicznymi. Dlatego zaufanie do lekarza jest kluczowe. I jeśli pacjent z jakiegoś powodu, czy to przez działania niepożądane, czy ze względów finansowych, zamierza przestać przyjmować leki, kluczowym jest, żeby nas o tym poinformował. Wówczas mamy szansę zareagować i zmodyfikować leczenie w taki sposób, żeby ciągłość leczenia jednak została zachowana. Obserwujemy pacjentów, u których rozpoznanie ustalono dzięki screeningowi rodzin, i nigdy nie mieli świadomości, do jakich szkód ta choroba może doprowadzić, bo leczenie zostało włączone wcześnie. Jeśli po latach z jakiegokolwiek powodu odstawiają leczenie i pojawiają się nieodwracalne objawy choroby, przychodzi przykra konstatacja zmarnowanej szansy.
Rozmawiał Jerzy Dziekoński