Rdzeniowy zanik mięśni (spinal muscular atrophy – SMA) jest jedną z częstszych i cięższych chorób uwarunkowanych genetycznie. Do niedawna był chorobą nieuleczalną, ale w ciągu ostatnich lat nastąpił przełom w leczeniu SMA i są leki, które mogą zatrzymać postęp choroby, a podane zanim objawy się rozwiną – zapobiec ich wystąpieniu. Od kwietnia 2021 roku w Polsce jest wdrażany program badań przesiewowych noworodków
Co to jest SMA i jakie są jego przyczyny?
Rdzeniowy zanik mięśni (ang. spinal muscular atrophy – SMA) jest chorobą genetycznie uwarunkowaną, dziedziczoną jako cecha autosomalna recesywna. W przebiegu choroby obumierają motoneurony rdzenia kręgowego (komórki nerwowe, za pośrednictwem których sygnały są przekazywane do mięśni). W konsekwencji dochodzi do osłabienia i zaniku niemal wszystkich grup mięśniowych. Nieleczony SMA jest chorobą szybko postępującą, prowadzącą do niewydolności oddechowej i porażenia czterokończynowego w postaciach ciężkich lub znacznej niepełnosprawności ruchowej w postaciach przewlekłych.
Objawy rdzeniowego zaniku mięśni mogą wystąpić w każdym wieku – od okresu płodowego po wiek dorosły. U większości pacjentów (ok. 90%) ujawniają się jednak do końca 2. roku życia. Wczesne wystąpienie objawów wiąże się z cięższym przebiegiem i poważniejszym rokowaniem.
Ze względu na zmienny wiek wystąpienia i nasilenia objawów klinicznych SMA wyróżnia się 5 postaci choroby:
- SMA0 – postać prenatalna, stanowi nie więcej niż 1–2% przypadków SMA,
- SMA1 – początek przed 6. mż.; odpowiada za 40–50% przypadków SMA
- SMA2 – początek w 6.–18. mż.; stanowi 20–30% przypadków SMA,
- SMA3 – początek objawów po osiągnięciu zdolności chodzenia, odpowiada za ok. 20–30% zachorowań na SMA,
- SMA4 – początek objawów w wieku dorosłym, zazwyczaj w 3. dekadzie życia, jest bardzo rzadka, odpowiada za 1–2% zachorowań na SMA
Podstawą tego podziału są wiek wystąpienia objawów i najlepszy osiągnięty krok milowy rozwoju ruchowego (siedzenie i chodzenie).
Jak często występuje rdzeniowy zanik mięśni?
SMA jest jedną z częstszych chorób genetycznie uwarunkowanych o zachorowalności porównywalnej z fenyloketonurią. W populacji europejskiej zapadalność na SMA szacuje się na około 1 : 8400 urodzeń (11,9/100 000). W latach 2011–2015 w Polsce SMA rozpoznano u 240 pacjentów, co odpowiada częstości zachorowania około 1 : 8300 urodzeń. Badania epidemiologiczne prowadzone w Polsce na początku pierwszej dekady dwudziestego wieku wskazywały na zachorowalność na poziomie 1 : 7500–9000. Szacuje się, że w Polsce żyje około 1000 pacjentów z SMA, a rocznie chorobę tę rozpoznaje się u około 50 osób.
Jak się objawia rdzeniowy zanik mięśni?
Głównymi objawami rdzeniowego zaniku mięśni jest symetryczne (po obu stronach ciała) osłabienie i zanik mięśni tułowia i kończyn. Czas wystąpienia objawów i ich nasilenie różni się w zależności od rodzaju SMA.
Objawy SMA u niemowląt
U dzieci z SMA0 objawy rozwijają się w wieku płodowym. Matka może odczuwać słabsze ruchy dziecka. Po urodzeniu dziecko jest wiotkie, ma zaburzenia ssania i połykania oraz osłabienie lub brak ruchów czynnych (samodzielnie wykonywanych, bez pomocy z zewnątrz), czemu mogą towarzyszyć przykurcze wielostawowe oraz cofnięta żuchwa. Szybko rozwija się niewydolność oddechowa. Rocznie w Polsce rodzi się prawdopodobnie jedno dziecko z tym typem choroby.
U dzieci z SMA1 okres okołoporodowy i noworodkowy przebiega zwykle bez powikłań. Pierwsze objawy rozpoznaje się najczęściej w 2. lub 3. miesiącu życia. Dziecko męczy się przy jedzeniu, cicho płacze, słabiej kopie nogami, nie dźwiga głowy. Wiotkość i osłabienie mięśni narastają szybko. Dziecko nie nabywa nowych umiejętności, coraz słabiej porusza nogami, słabiej przybiera masę ciała, nawracają infekcje dróg oddechowych, widoczny jest wysiłek oddechowy. Jednoczenie mimika twarzy jest zachowana, podobnie jak bardzo dobry kontakt emocjonalny. Choroba nieleczona prowadzi do rozwoju niewydolności oddechowej. Bez leczenia do 2. roku życia większość dzieci umiera lub staje się w pełni zależna od respiratora i nie osiąga zdolności samodzielnego siedzenia.
W postaci SMA2 objawy pojawiają się między 6. i 18. miesiącem życia. W pierwszych kilku miesiącach życia rozwój wydaje się prawidłowy. Dziecko osiąga zdolność samodzielnego siedzenia w oczekiwanym wieku lub z nieznacznym opóźnieniem. Niektóre dzieci zaczynają raczkować i wstawać przy sprzętach. Następnie rozwój ruchowy zostaje jednak zahamowany, dziecko słabnie i nie osiąga zdolności samodzielnego chodu. Charakterystyczne dla tej postaci są drżenie palców oraz bardzo wiotkie dłonie. Szybko dochodzi do powikłań w układzie ruchu – narastają przykurcze wielostawowe, wcześnie rozwija się skolioza. Większość pacjentów z czasem wymaga nieinwazyjnego wsparcia oddechu. Stopień niepełnosprawności ruchowej jest bardzo duży. Rozwój intelektualny jest prawidłowy.
Postać SMA3 jest najbardziej zróżnicowana w swoim przebiegu. Objawy osłabienia siły mięśniowej pojawiają się po osiągnieciu zdolności samodzielnego chodzenia, tzn. między 18. miesiącem życia a 18. rokiem życia. Zazwyczaj im późniejszy początek choroby, tym przebiega ona łagodniej, ale niezależnie od wieku wystąpienia pierwszych objawów, zaburzenia ruchowe nieuchronnie postępują. Większość pacjentów, którzy zachorowali przed 3. rokiem życia, traci zdolność samodzielnego chodzenia w okresie dziecięcym lub nastoletnim.
W przypadku SMA4 pierwsze objawy pojawiają się w wieku dorosłym. W początkowym okresie dominuje osłabienie mięśni obręczy biodrowej. Pacjent ma trudności ze wstawaniem z pozycji kucznej, bieganiem i wchodzeniem po schodach. Podobnie jak SMA0 jest to niezwykle rzadka forma choroby.
Jak lekarz stawia diagnozę rdzeniowego zaniku mięśni?
SMA – badania przesiewowe
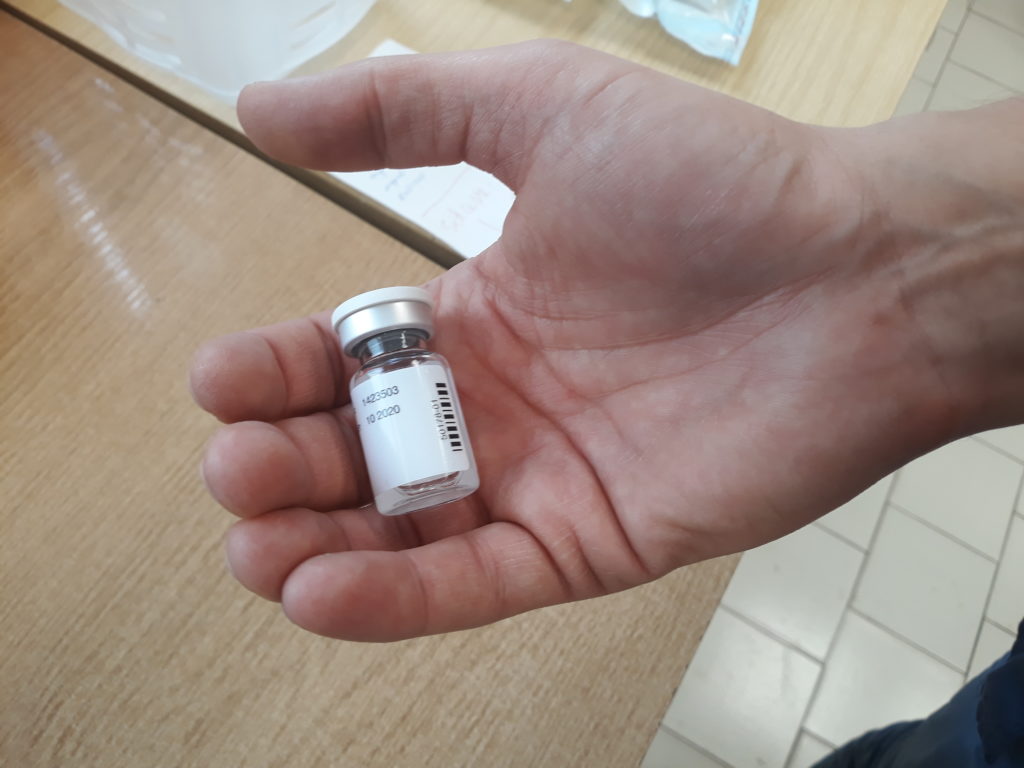
W lutym 2021 r. do Programu Badań Noworodków w Rzeczypospolitej Polskiej na lata 2019–2022 włączono badanie przesiewowe w kierunku rdzeniowego zaniku mięśni (SMA). Wykonuje się je z próbek krwi pobieranych rutynowo od noworodków na panel przesiewowy 30 chorób wrodzonych. Schemat postępowania przesiewowego zapewnia szybką ścieżkę od pobrania krwi dziecka do decyzji Komisji ds. Leczenia SMA przy Ministerstwie Zdrowia w przypadku, jeśli okaże się, że dziecko jest chore.
W każdym województwie znajduje się wyznaczony ośrodek referencyjny odpowiedzialny za leczenie SMA. Zakodowany wynik dodatniego wyniku testu w kierunku SMA otrzymuje neurolog z ośrodka referencyjnego w województwie właściwym dla miejsca zamieszkania dziecka. Specjalista neurolog w ośrodku referencyjnym jest odpowiedzialny za pilne wezwanie noworodka, uzyskanie zgody rodziców na badanie molekularne w kierunku SMA, pobranie próbki krwi żylnej na badanie weryfikujące i wysłanie jej w ciągu 24 godzin do Zakładu Genetyki Medycznej IMiD. Zweryfikowany wynik badania, jest przekazywany bezzwłocznie do lekarza prowadzącego oraz do Komisji ds. Leczenia SMA przy Ministerstwie Zdrowia.
Program badań przesiewowych w kierunku SMA jest wdrażany stopniowo w kolejnych województwach od kwietnia 2021 roku, a do końca 2022 roku ma objąć cały kraj. Możliwe są jednak korekty przyspieszające wdrożenie badań w ostatnich województwach. Plan na 2022 rok zakłada zbadanie 270 000 dzieci, czyli około 80% populacji noworodków.
W przypadku osób, których nie objęło badanie przesiewowe po urodzeniu przy podejrzeniu SMA lekarz najpierw zbiera wywiad od pacjenta lub jego rodziny oraz bada pacjenta, a następnie kieruje do neurologa, który może zlecić badania dodatkowe, takie jak elektromiografia, czyli badanie czynności mięśni. Ostateczne rozpoznanie ustala się na podstawie wyniku badania genetycznego z krwi pobranej od pacjenta.
Jakie są sposoby leczenia rdzeniowego zaniku mięśni?
Do niedawna SMA był chorobą nieuleczalną, prowadzącą do niewydolności oddechowej, a także ciężkiej niepełnosprawności ruchowej. W ostatnich latach nastąpił przełom w leczeniu SMA. Warunkiem skuteczności leczenia SMA jest wczesne rozpoczęcie terapii, zanim dojdzie do nieodwracalnego zaniku motoneuronów rdzenia kręgowego Zarejestrowano trzy leki o działaniu przyczynowym, zwiększające ilość białka SMN (survival of motor neuron), którego niedobór jest przyczyną choroby: nusinersen, onasemnogen abeparwowek i rysdyplam. Wszystkie wymienione leki wykazują dużą skuteczność, uzależnioną jednak od stopnia zaawansowania choroby w momencie rozpoczęcia terapii. Najlepsze wyniki uzyskuje się, rozpoczynając leczenie w stadium przedobjawowym.
Od 2019 r. dostępne jest w Polsce bezpłatnie leczenie nusinersenem dla wszystkich chorych na rdzeniowy zanik mięśni, objawowych i bezobjawowych, dzieci i dorosłych, w ramach programu lekowego Narodowego Funduszu Zdrowia.
W lutym 2021 r. w wykazie technologii medycznych o wysokim poziomie innowacyjności Agencji Oceny Technologii Medycznych i Taryfikacji znalazł się preparat Zolgensma (onasemnogen abeparwowek), co daje nadzieję na uruchomienie w niedalekiej przyszłości programu refundacji również tego leku.
Czy możliwe jest całkowite wyleczenie rdzeniowego zaniku mięśni?
Najważniejsze jest jak najwcześniejsze rozpoczęcie leczenia. Jeśli leczenie SMA rozpocznie się zanim wystąpią pierwsze objawy, jest możliwe, że pacjent będzie rozwijał się podobnie do swoich zdrowych rówieśników.