Przyczyną opóźnionego rozpoznania SMA jest późne pojawienie się objawów i niedostrzeżenie przez lekarza, który bada dziecko, pierwszych sygnałów osłabienia mięśniowego – mówi prof. dr hab. n. med. Maria Mazurkiewicz-Bełdzińska, przewodnicząca Polskiego Towarzystwa Neurologów Dziecięcych, kierownik Kliniki Neurologii Rozwojowej Gdańskiego Uniwersytetu Medycznego.

Prof. dr hab. n. med. Maria Mazurkiewicz-Bełdzińska
Katarzyna Pelc: W 2021 roku wprowadzono w Polsce program badań przesiewowych w kierunku rdzeniowego zaniku mięśni (spinal muscular atrophy – SMA). W przyszłym roku ma objąć już wszystkie dzieci urodzone w Polsce. Jaki materiał pobiera się do tego badania? Co się analizuje? Czy wynik będzie podlegał dodatkowej weryfikacji?
Prof. dr hab. n. med. Maria Mazurkiewicz-Bełdzińska: Badanie przesiewowe w kierunku SMA wykonuje się tuż po urodzeniu z kropli krwi pobranej od noworodka na specjalną bibułę, którą następnie przesyła się do jednego z siedmiu laboratoriów w kraju. Analizuje się występowanie mutacji w obrębie genu SMN1 na długim ramieniu chromosomu 5 (stąd tę postać SMA nazywa się SMA 5q). W przypadku wykrycia choroby zleca się badanie weryfikacyjne. Jednocześnie ocenia się liczbę zapasowych kopii genu SMN2. Wynik powinien być dostępny w ciągu kilku dni. To pozwala określić, z jakim potencjalnym obrazem klinicznym choroby możemy mieć do czynienia u konkretnego dziecka. Jeśli dziecko ma 1–2 zapasowe kopie tego genu, czyli może się rozwinąć postać ciężka, leczenie należy rozpocząć natychmiast po ustaleniu rozpoznania.
Jaka jest czułość i swoistość metod przesiewowych? Jakie jest ryzyko pominięcia chorego dziecka lub nierozpoznania choroby?
To >95%. Badanie nie wychwytuje mutacji punktowych. Ryzyko pominięcia lub nierozpoznania choroby jest bardzo niewielkie.
Ile szacunkowo rodzi się w Polsce dzieci z SMA?
Jest to bardzo rzadka choroba, liczymy, że rocznie może się z nią urodzić około 50 dzieci.
Czy na świecie są prowadzone takie programy badań przesiewowych?
Jesteśmy pionierami, jednymi z pierwszych. Od tego roku badania takie są też prowadzone w Holandii, Niemczech, Serbii i Słowenii, a od wiosny także w poszczególnych regionach Francji (od 2023 r. mają objąć cały kraj). W Stanach Zjednoczonych badanych jest około 63% noworodków, w Kanadzie – badania przesiewowe obejmują wszystkie dzieci urodzone w prowincji Quebec, a w Australii – noworodki z terenu Terytorium Stołecznego oraz Nowej Południowej Walii. W Rosji i we Włoszech trwają programy pilotażowe, a w Hiszpanii i Wielkiej Brytanii są w trakcie przygotowań. W Austrii, Norwegii i Szwecji rozpatrywane są wnioski o wdrożenie narodowego przesiewu w kierunku SMA.
Czy poprawiło to rokowanie u dzieci z SMA?
To zupełnie zrozumiałe, że badania te przyspieszą leczenie, a tym samym poprawią rokowania. Mamy badania obejmujące pacjentów bezobjawowych i widać znakomicie, że są to pacjenci, którzy osiągają zdrowe fenotypy.
Jaka jest przyczyna SMA?
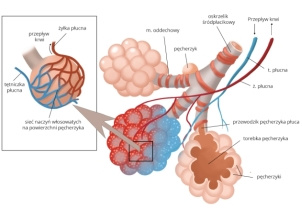
SMA to ciężka i rzadka choroba, w której z powodu wady genetycznej obumierają neurony w rdzeniu kręgowym odpowiadające za skurcze i rozkurcze mięśni. Brak impulsów nerwowych
prowadzi do uogólnionego osłabienia i postępującego
zaniku mięśni szkieletowych, a w ostateczności
– do częściowego albo całkowitego paraliżu.
Choroba dotyka osób w różnym wieku, jednak w >90% przypadków objawy pojawiają się w niemowlęctwie
albo we wczesnym dzieciństwie. SMA
jest wynikiem wady genetycznej – mutacji w genie
SMN1, którą chory otrzymuje od obojga rodziców,
zazwyczaj nieświadomych jej posiadania.
Do czasu wprowadzenia nowoczesnych metod
opieki oddechowej i leczenia farmakologicznego
SMA była najczęstszą genetyczną przyczyną
śmierci dzieci do 2. roku życia. W Polsce w 2019
roku wprowadzono nowoczesne leczenie SMA, w pierwszej kolejności u wszystkich dzieci, co sprawiło,
że choroba nie jest już śmiertelna.
Tradycyjnie stosuje się podział na trzy postacie
(typy) SMA, w zależności od czasu wystąpienia
pierwszych objawów i osiągniętych etapów rozwoju
ruchowego.
W postaci pierwszej SMA, zwanej dawniej
„chorobą Werdniga i Hoffmanna”, objawy osłabienia
mięśni widoczne są w pierwszych tygodniach
albo miesiącach życia. Niemowlę jest wiotkie, zwykle
ma osłabiony krzyk i oddech, nie jest w stanie
unieść główki i nigdy nie będzie siedzieć bez podparcia.
Aby przeżyć, dzieci takie wymagają wysoko
wyspecjalizowanej opieki, a i tak śmiertelność w tej grupie jest duża.
Dzieci, które umiały utrzymać się bez podparcia w pozycji siedzącej, ale nie zaczęły chodzić
samodzielnie, zaliczane są do postaci drugiej
SMA. Również te dzieci potrzebują specjalistycznej
opieki i wsparcia, chociaż ryzyko przedwczesnej
śmierci jest w tej grupie znacznie mniejsze.
Postać trzecią SMA, dawniej zwaną „chorobą
Kugelberga i Welander”, rozpoznaje się u osób, które
były w stanie samodzielnie postawić co najmniej
kilka kroków, zanim osłabienie mięśni zmusiło je
do korzystania z wózka. Osoby z tym typem SMA
zwykle prowadzą w miarę niezależne życie, często
jednak muszą korzystać ze specjalistycznego
sprzętu.
W przypadku wystąpienia pierwszych objawów w wieku dorosłym niektórzy lekarze rozpoznają
czwartą postać SMA. W tej postaci mięśnie są
osłabione w najmniejszym zakresie.
Od czego zależy ciężkość przebiegu choroby?
Z patogenezą SMA związane są dwa bliźniaczo
podobne geny – SMN1 i SMN2. Za występowanie
objawów choroby odpowiedzialne są mutacje genu
SMN1, natomiast gen SMN2 pełni rolę modyfikatora
fenotypu. Mutacje genu SMN2 nie są patogenne.
Natomiast zwiększenie liczby kopii genu
SMN2 na skutek duplikacji czy konwersji łagodzi
przebieg kliniczny SMA.
Badania wykazały silną korelację między liczbą
kopii SMN2 a fenotypem choroby. Pacjenci z formą
ciężką mają zwykle 1–2 kopii, z postacią pośrednią
2–3, a z formą łagodną 3–4, a nawet 5 i 6 kopii.
Nie wszystkie dzieci zdiagnozowano wkrótce po urodzeniu. Jakie są najczęstsze przyczyny opóźnionego rozpoznania?
Przyczyną opóźnionego rozpoznania jest późne pojawienie się objawów i niedostrzeżenie przez lekarza, który bada dziecko, pierwszych sygnałów osłabienia mięśniowego. Pierwszy typ SMA jest łatwiej rozpoznać, bo objawy są cięższe, a pozostałe niekiedy trudno jest rozróżnić.
Co mogą zrobić pediatrzy i lekarze rodzinni, aby wyeliminować takie opóźnienia?
Teraz mamy już przesiew i to on powinien wyeliminować wszelkie opóźnienia. Koniecznie trzeba jednak zachować czujność, zwłaszcza ze względu na pacjentów z mutacjami punktowymi, którzy potencjalnie nie są wykrywani w badaniach przesiewowych. Ważne jest, aby lekarze powszechnie znali objawy SMA.
Jaki jest naturalny przebieg nieleczonej choroby?
Śmierć do 2. roku życia w pierwszej postaci. W drugiej postaci pacjent nigdy nie będzie chodził, pojawią się głębokie skoliozy, bardzo duże powikłania dotyczące wad postawy, mogą się pojawić powikłania oddechowe i trawienne. W postaci trzeciej głębokie osłabienie siły mięśniowej i ruchów dowolnych, w wyniku których pacjent przestaje chodzić.
Jakimi metodami leczenia dysponujemy współcześnie?
Nusinersen (nazwa handlowa Spinraza®) to
pierwszy na świecie lek opracowany w celu przyczynowego
leczenia SMA. Jest stosowany od 2016
roku (w Unii Europejskiej od 2017 r.). Na świecie
przyjmuje go kilkanaście tysięcy osób, a jego działanie
jest dobrze znane.
Onasemnogen abeparwowek (nazwa handlowa
Zolgensma®) jest innowacyjnym lekiem opracowanym w celu leczenia SMA. Na rynku dostępny jest
od 2019 roku, a w Unii Europejskiej od maja 2020
roku. Zolgensma należy do tzw. terapii genowych.
Rysdyplam (nazwa handlowa: Evrysdi™) jest
lekiem opracowanym do przyczynowego leczenia
SMA. Ma postać syropu, który przyjmuje się doustnie
raz dziennie (ew. przez sondę albo tzw. PEG).
Rysdyplam jest zarejestrowany w Stanach Zjednoczonych od sierpnia 2019 roku, a w Unii Europejskiej
(w tym w Polsce) od marca 2021 roku;
ponadto ma rejestrację w szeregu innych państw.
Które z nich są dostępne oraz refundowane w Polsce?
Od 1 stycznia 2019 roku minister zdrowia objął refundacją lek Spinraza (nusinersen) w ramach programu lekowego „Leczenie rdzeniowego zaniku mięśni” u przedobjawowych i objawowych pacjentów z rozpoznaniem SMA 5q potwierdzonym w badaniach genetycznych. Jest to bardzo szerokie kryterium refundacji, wyróżniające Polskę nawet na tle państw europejskich. Nie ma ograniczeń w zakresie rodzaju choroby, wieku i parametrów płucnych (wentylacyjnych) – jest to najszerszy możliwy przekrój populacji, jaki można było objąć refundacją.
Od ponad roku wszyscy chorzy mają dostęp
do bezpłatnego leczenia o udowodnionej skuteczności.
Program lekowy działa i na bieżąco są
do niego włączani pacjenci potrzebujący przedmiotowej
terapii. Proces ten stale nadzoruje też
Zespół Koordynacyjny. Wciąż zwiększa się liczba
pacjentów uczestniczących w terapii – wnioski
szpitali o rozpoczęcie terapii u nowych chorych
są rozpatrywane na bieżąco. Do systemu monitorowania
programu terapeutycznego wpisano już
>500 pacjentów, a wyniki wydają się być obiecujące z punktu widzenia klinicznego.
Zolgensma, czyli terapia genowa, do tej pory nie
jest refundowana przez NFZ. Znalazła się jednak
na liście Funduszu Medycznego i podejmowane są
starania dotyczące jej dostępności, tak aby chorzy i lekarze mieli możliwość wyboru optymalnej metody
leczenia, zwłaszcza u najmłodszych dzieci, u których leczenie genowe jest wyjątkowo skuteczne.
Poza Stanami Zjednoczonymi, lek Zolgensma
jest objęty refundacją przez system publicznej
opieki zdrowotnej w Austrii (z limitem rocznym i ograniczeniami), Brazylii (zgodnie ze wskazaniami
rejestracyjnymi), Czechach (z ograniczeniami),
we Francji (z ograniczeniami), w Izraelu (zgodnie
ze wskazaniami rejestracyjnymi), Japonii,
Katarze (zgodnie ze wskazaniami rejestracyjnymi,
ale tylko dla obywateli Kataru), Niemczech
(z ograniczeniami), na Słowacji (z ograniczeniami), w Szwajcarii (z ograniczeniami, na zasadzie wczesnego
dostępu) i we Włoszech (z ograniczeniami).
Rysdyplam na razie nie jest refundowany.
Od 2020 roku producent nieodpłatnie dostarcza
ten lek chorym na najcięższą postać choroby w ramach globalnego programu dostępu. W Polsce
do programu zakwalifikowano 50 pacjentów. Nabór
do programu jest już zamknięty.