Hipercholesterolemia rodzinna to uwarunkowana genetycznie choroba, która powoduje zwiększone stężenie cholesterolu oraz związane z tym powikłania (choroby układu sercowo-naczyniowego, nawet w bardzo młodym wieku). Leczenie jest takie, jak w przypadku innych rodzajów hipercholesterolemii, polega na modyfikacji stylu życia oraz przyjmowaniu leków.
Co to jest hipercholesterolemia rodzinna i jakie są jej przyczyny?
Rodzinna hipercholesterolemia (heterozygous familial hypercholesterolemia – HeFH) to choroba uwarunkowana genetycznie, która powoduje zwiększenie stężenia we krwi zarówno całkowitego cholesterolu, jak i lipoprotein o małej gęstości (low density lipoprotein – LDL), czyli tak zwanego „złego cholesterolu”.
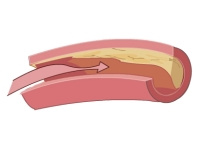
Cholesterol jest lipidem pełniącym ważne funkcje w organizmie. Jego pochodne wchodzą w skład błon komórkowych, bierze on też udział w wielu procesach metabolicznych. Jego nadmiar może jednak być szkodliwy. Kiedy jest wbudowywany w ścianę naczyń krwionośnych, prowadzi do niebezpiecznej choroby, jaką jest miażdżyca. Dlatego duże stężenie cholesterolu jest czynnikiem ryzyka choroby niedokrwiennej serca i zawału serca (czytaj więcej o cholesterolu).
Osoby obciążone chorobą genetyczną, jaką jest hipercholesterolemia rodzinna, mają zdecydowanie większe stężenie cholesterolu niż osoby bez tej mutacji genetycznej. Ich problemy z hipercholesterolemią zaczynają się już we wczesnym dzieciństwie, a powikłania w postaci chorób serca i naczyń mogą się pojawić w młodym wieku. U osób z hipercholesterolemią rodzinną już w wieku około 45 lat mogą występować zaburzenia porównywalne ze stwierdzanymi przeciętnie w wieku 70 lat. Dlatego chorzy z hipercholesterolemią rodzinną powinni pozostawać pod regularną opieką lekarską i stosować odpowiednie leczenie mające na celu zmniejszenie stężenia cholesterolu i LDL.
Prawidłowo wątroba wychwytuje cząstki LDL z krwi. Mutacja genu receptora LDL (LDLR), rzadziej mutacja genu apolipoproteiny B (apoB) czy PCSK9 (proprotein convertase subtilisin/kexin type 9) odpowiedzialne za występowanie rodzinnej hipercholesterolemii, sprawiają, iż krew nie może być prawidłowo, efektywnie oczyszczana z krążących cząsteczek LDL. W efekcie „zły cholesterol” gromadzi się, odkłada w ścianach naczyń oraz innych tkankach, co prowadzi do niebezpiecznych zaburzeń.
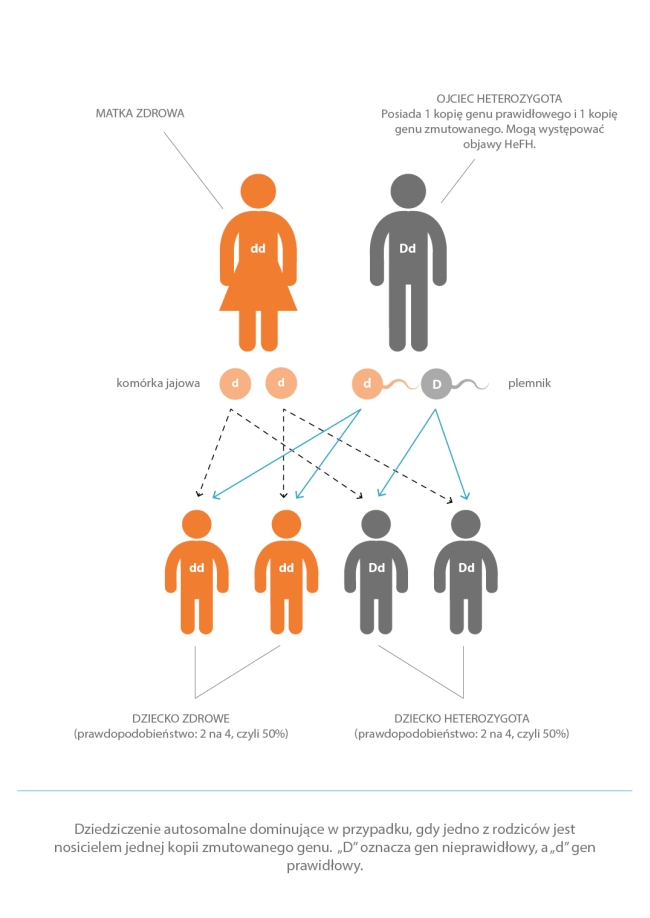
Rodzinna hipercholesterolemia to choroba genetyczna dziedziczona w sposób autosomalny dominujący. To oznacza, że jest to choroba, którą można odziedziczyć po jednym z rodziców. Opisano wiele różnych mutacji prowadzących do choroby. Zmutowany gen położony jest na 19. chromosomie. Każdy człowiek ma po dwie kopie każdego genu. Zarówno ojciec, jak i matka przekazuje swojemu potomstwu po jednej kopii. Jeśli jedno z rodziców ma jeden nieprawidłowy gen odpowiedzialny za rodzinną hipercholesterolemię, istnieje 50% ryzyko, że ich dziecko także będzie obciążone chorobą, gdyż wystarczy jedna nieprawidłowa kopia genu – odziedziczona tylko od matki albo tylko od ojca.
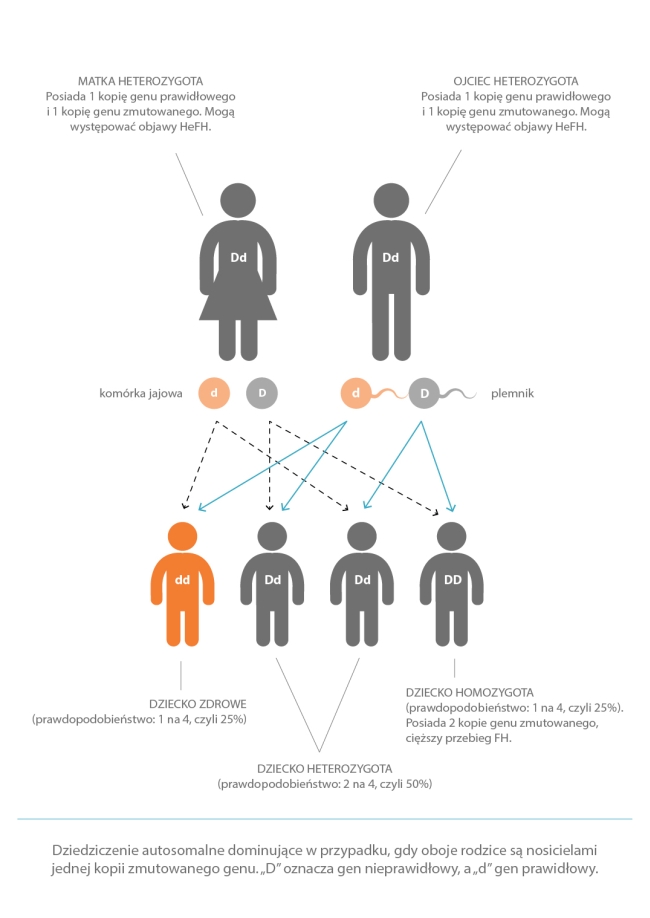
Jeśli zdarzy się, że oboje rodzice chorują, ryzyko zachorowania jest odpowiednio większe (łącznie 75%). Może się tak zdarzyć, że oboje rodzice przekażą dziecku nieprawidłową kopię genu. Wówczas nasilenie choroby u dziecka jest większe, niż w przypadku mutacji odziedziczonej tylko po jednym z rodziców. Osoba z jedną nieprawidłową kopią genu jest określana jako heterozygota. Osoba z dwiema nieprawidłowymi kopiami genu nazywana jest homozygotą. Ma jeszcze większe stężenie cholesterolu, a problemy zdrowotne, takie jak miażdżyca, zaczynają się u niej już we wczesnym dzieciństwie. Osoby te mają też zwiększone ryzyko zgonu w młodym wieku. Jeśli któreś z rodziców jest homozygotą, czyli ma obie nieprawidłowe kopie genu, wówczas każde jego dziecko będzie obarczone mutacją.
Jak się objawia rodzinna hipercholesterolemia?
Duże stężenie cholesterolu u osoby z rodzinną hipercholesterolemią będącej heterozygotą (z jedną nieprawidłową kopią genu) z reguły długo nie daje żadnych objawów odczuwanych przez pacjenta. Można by pomyśleć, że skoro choroba nie powoduje dolegliwości, nie jest poważnym problemem. W rzeczywistości jest to jednak niebezpieczna sytuacja, gdyż szkody w organizmie powstają bez wiedzy osoby chorej. Dolegliwości są zwykle efektem długotrwałego narastania powikłań wynikających z hipercholesterolemii. Są to przede wszystkim:
- Bóle dławicowe w klatce piersiowej – rozlany ból, opisywany często jako uczucie „dławienia”, pojawiający się najczęściej po wysiłku jest objawem miażdżycy naczyń wieńcowych i choroby niedokrwiennej serca. Zmiany miażdżycowe w naczyniach wieńcowych, prowadzące do choroby niedokrwiennej serca obecne są już nawet u chorych w wieku 11–23 lat w przypadku osób z jedną nieprawidłową kopią genu.
- W przebiegu miażdżycy może dojść do zwężenia naczyń i niedokrwienia różnych obszarów i narządów, nie tylko serca. Niedokrwienie kończyn dolnych daje objawy chromania przestankowego, czyli bólów nóg pojawiających się początkowo po wysiłku (chodzenie, bieganie), a w zaawansowanych postaciach także w spoczynku. Niedokrwienie w obszarze kończyn górnych może dawać analogiczne objawy w rękach (czyli np. ból pojawiający się po noszeniu ciężarów). Dolegliwościom ze strony ręki mogą towarzyszyć objawy neurologiczne. Miażdżyca tętnic szyjnych prowadzi do objawów neurologicznych, czyli zawrotów głowy, zasłabnięć, a także wiąże się z ryzykiem udaru niedokrwiennego. Niedokrwienie jelit wywołuje bóle brzucha, początkowo po posiłkach, a w zaawansowanych przypadkach może prowadzić do wysoce śmiertelnego powikłania, jakim jest zawał krezki.
- Nawracające zapalenia i dolegliwości bólowe ze strony ścięgna Achillesa, rzadziej innych ścięgien.
- Kępki żółte, czyli złogi cholesterolu odkładające się na skórze łokci, kolan, pośladków, na ścięgnach i wokół oczu. Zmiany skórne, to nagle pojawiające się żółte bądź żółtobrunatne kropki na skórze z czerwoną obwódką – najczęściej lokalizują się w tej postaci na pośladkach, z tyłu na udach, na łokciach i kolanach. Zmiany mogą mieć także charakter nieco większych guzków skórnych, nazywanych żółtakami, które najczęściej lokalizują się nad dużymi stawami i pojawiają się stopniowo. Żółtaki powstają z wiekiem, ostatecznie ujawniają się u około 75% osób z jedną kopią nieprawidłowego genu. Guzki powstałe ze złogów cholesterolu mogą się także lokalizować na ścięgnach oraz strukturach otaczających kość. Najczęściej obecne są w ścięgnie Achillesa (nad piętą) oraz na grzbiecie dłoni. Mogą działać drażniąco i wyzwalać proces zapalny odpowiedzialny za wspomniane, nawracające zapalenia i ból w obrębie ścięgna Achillesa. Żółtaki powiek to depozyty cholesterolu w tkankach okolicy oczu.
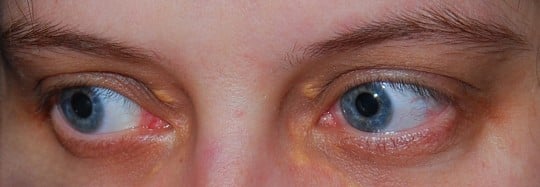 Kępki żółte" />
Kępki żółte" />
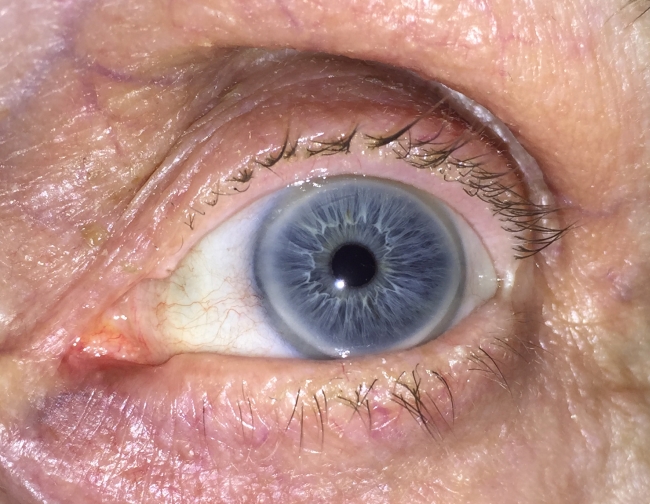
- Rąbek starczy rogówki – biaława „obwódka starcza” na oku, powstała w wyniku kumulacji lipidów (czyli tłuszczów) w obrębie struktur oka.
Jak często występuje rodzinna hipercholesterolemia?
Rodzinna hipercholesterolemia to choroba stosunkowo częsta. Jest to także najczęstsza spośród genetycznie uwarunkowanych postaci hipercholesterolemii. Szacuje się, że w populacji polskiej 1/250 osób w wieku 20–74 lat osób ma nieprawidłowy gen odpowiedzialny za hipercholesterolemię (jest heterozygotą), co oznacza, że na rodzinną hipercholesterolemię choruje 0,4% wszystkich osób w populacji ogólnej.
U około 1% osób z hipercholesterolemią występuje hipercholesterolemia rodzinna. U pozostałych, duże stężenie cholesterolu wynika z innych przyczyn, najczęściej z niezdrowego stylu życia, cukrzycy, czy innych chorób, w tym niektórych zaburzeń hormonalnych.
Szacuje się, że na świecie żyje około 10 milionów osób z rodzinną hipercholesterolemią. 40% z nich nie wie o swojej chorobie, a połowa nie jest leczona. W Polsce choruje około 80 000 osób, ale jedynie znikomy odsetek jest świadomy swojej choroby.
Homozygotyczna hipercholesterolemia rodzinna, która występuje u osób z dwoma nieprawidłowymi kopiami genu zdarza się u 1 osoby na milion.
W niektórych populacjach występowanie mutacji (heterozygotyczna rodzinna hipercholesterolemia) jest częstsze niż w Polsce. Do grupy zwiększonego ryzyka choroby należą osoby mające korzenie w wymienionych populacjach:
- mieszkańcy kanadyjskiej prowincji Quebec pochodzenia francuskiego – 1:270 osób
- libańscy chrześcijanie – 1:170 osób
- afrykanerzy – 1:100 osób
- Żydzi aszkenazyjscy – 1:67 osób
Co robić w przypadku wystąpienia objawów rodzinnej hipercholesterolemii?
W razie pojawienia się opisanych powyżej dolegliwości czy żółtaków należy zgłosić się do lekarza podstawowej opieki zdrowotnej. Dokładna rozmowa na temat objawów i czynników ryzyka oraz badanie lekarskie pozwolą ocenić, czy problemy wynikają z rodzinnej hipercholesterolemii i niedokrwienia na tle miażdżycy, czy istnieje inna, bardziej prawdopodobna przyczyna dolegliwości.
Z reguły lekarz podstawowej opieki zdrowotnej kieruje pacjenta na podstawowe badania dodatkowe, w tym badanie profilu lipidów we krwi (tzn. ocenę stężeń cholesterolu całkowitego, LDL, HDL i triglicerydów). W zależności od wyników badań oraz lokalizacji zmian, pacjenci z podejrzeniem rodzinnej hipercholesterolemii otrzymują od lekarza POZ wstępne rozpoznanie i zalecenia terapeutyczne. Następnie są kierowani do specjalistów – kardiologa, angiologa czy chirurga naczyniowego, a w uzasadnionych przypadkach do poradni genetycznej, w celu poszerzenia diagnostyki oraz doboru optymalnej terapii.
Osoby z rodzinną hipercholesterolemią, które pozostają nieleczone, są w większym stopniu zagrożone chorobami układu sercowo-naczyniowego, w tym zawałem serca i udarem mózgu. Dodatkowo do zdarzeń takich może u nich dojść w stosunkowo młodym wieku.
Wymienione poniżej objawy mogą świadczyć o nagłym pogorszeniu niedokrwienia, a nawet całkowitym zamknięciu dopływu krwi do narządów. Są to sytuacje stwarzające bezpośrednie zagrożenie dla życia, dlatego wymagają natychmiastowego wezwania pogotowia ratunkowego lub zgłoszenia się na szpitalny oddział ratunkowy:
- nagłe wystąpienie silnego bólu w klatce piersiowej, szczególnie utrzymującego się pomimo spoczynku, a nawet po przyjęciu nitrogliceryny
- nagłe zasłabnięcia i utrata przytomności
- nagłe objawy niedowładów, opadnięcie kącika ust, zaburzenia mowy, zaburzenia równowagi
- silny ból brzucha z zatrzymaniem perystaltyki (zaparcie, brak oddawania gazów, wymioty)
- silny ból, zblednięcie i oziębienie kończyny dolnej (częściej) lub górnej, z czasem pojawiają się także zaburzenia czucia oraz porażenie nogi lub ręki.
Stopniowe pogarszanie tolerancji wysiłku, ograniczanego dolegliwościami bólowymi, czyli np. skracanie dystansu chromania przestankowego, czyli odcinka, jaki jest się w stanie przejść bez bólu w nogach bądź pojawienie się bólu nawet w spoczynku, świadczy o tym, że choroba wynikająca z hipercholesterolemii postępuje. Taka sytuacja wymaga niezwłocznej konsultacji lekarskiej.
Osoby ze zdiagnozowaną rodzinną hipercholesterolemią powinny być świadome tego, iż choroba z dużym prawdopodobieństwem może zostać przekazana ich potomstwu. Dlatego powinny od samego początku przestrzegać zasad zdrowego stylu życia swojego dziecka. Pediatrę należy poinformować o obciążeniu rodzinnym chorobą oraz regularnie (według zaleceń pediatry) wykonywać badania kontrolne lipidów we krwi u dziecka. Pediatra kieruje dziecko na ewentualne dodatkowe badania. Szybkie zdiagnozowanie choroby u dziecka pozwala na odpowiednio wczesne wdrożenie zasad profilaktyki i terapii. Dzięki temu, pomimo mutacji genu, dziecko może uniknąć powikłań choroby.
W jaki sposób lekarz ustala rozpoznanie rodzinnej hipercholesterolemii?
Z reguły istnieje kilka scenariuszy, prowadzących do podjęcia diagnostyki rodzinnej hipercholesterolemii:
- diagnostyka rozpoczyna się już w wieku dziecięcym, gdyż wiadomo, iż któreś z rodziców (bądź oboje) chorują na rodzinną hipercholesterolemię
- diagnostyka rozpoczyna się od przypadkowego stwierdzenia w wynikach badań laboratoryjnych zwiększonego stężenia cholesterolu całkowitego i LDL
- pojawiają się wymienione wyżej objawy, świadczące o długotrwałym procesie chorobowym. Jest to najmniej korzystna, spośród wymienionych opcji, gdyż pacjent udaje się do lekarza na etapie, kiedy doszło już do sporych zaburzeń w organizmie. Oczywiście nadal można i należy zapobiegać dalszym powikłaniom.
Rozpoznanie rodzinnej hipercholesterolemii można w niektórych przypadkach oprzeć na danych z wywiadu oraz wynikach badania lekarskiego i badań dodatkowych, a szczególnie stężeniu cholesterolu całkowitego i LDL.
Stężenie cholesterolu całkowitego u osób z rodzinną hipercholesterolemią wynosi:
- ponad 250 mg/dl u dzieci
- ponad 300 mg/dl u dorosłych.
Stężenie LDL wynosi u tych osób:
- ponad 170-200 mg/dl u dzieci
- ponad 220 mg/dl u dorosłych.
Lekarz sprawdza też stężenie triglicerydów, które w przypadku rodzinnej hipercholesterolemii jest prawidłowe (tj. poniżej 150 mg/dl). Zwiększone stężenie triglicerydów może występować w innych postaciach zaburzeń lipidowych uwarunkowanych genetycznie.
W szacowaniu prawdopodobieństwa, z jakim zaburzenia lipidowe u pacjenta mogą wynikać z rodzinnej hipercholesterolemii bierze się pod uwagę kilka istotnych informacji, takich jak:
- obciążenie rodzinne, czyli:
- wczesne (przed 55. rokiem życia u mężczyzn, i przed 60. rokiem życia u kobiet) wystąpienie choroby wieńcowej w najbliższej rodzinie (pod pojęciem najbliższej rodziny należy rozumieć: rodziców, rodzeństwo, dzieci)
- inne choroby i stany wskazujące na taki wywiad u bliskich (choroba niedokrwienna serca, miażdżyca naczyń wieńcowych, ostry zespół wieńcowy, zawał serca, stan po przebytym zabiegu angioplastyki, wszczepieniu stentów w naczyniach wieńcowych bądź stan po wszczepieniu pomostów aortalno-wieńcowych)
- żółtaki ścięgien u najbliższych krewnych
- zwiększone stężenie LDL u najbliższych krewnych
- wczesne (przed 55. rokiem życia u mężczyzn, i przed 60. rokiem życia u kobiet) wystąpienie choroby wieńcowej lub choroby tętnic obwodowych (m.in. tętnic mózgu, tętnic kończyn górnych lub dolnych, jelit) u pacjenta
- żółtaki ścięgien u pacjenta
- rąbek starczy rogówki obecny przed 45. rokiem życia u pacjenta
- zbyt duże stężenie LDL we krwi – im wyższe, tym większe ryzyko podłoża rodzinnego (genetycznego) zaburzeń.
Należy wykluczyć inne możliwe przyczyny zaburzeń lipidowych, takie jak choroby nerek, cukrzyca, niedoczynność tarczycy, choroby wątroby, ciąża, stosowanie leków, które mogą powodować takie zaburzenia, w tym progestagenów, steroidów anabolicznych czy glikokortykosteroidów.
W diagnostyce rodzinnej hipercholesterolemii u osób z prawdopodobną chorobą wykorzystuje się także badania genetyczne (z pobranej od pacjenta próbki krwi). Jest to jedyny pewny test potwierdzający rozpoznanie. Wykonuje się także testy fibroblastów i komórek tkanki łącznej, oceniające zdolność „oczyszczania krwi” z LDL. W razie wykazania mutacji genetycznej odpowiedzialnej za rodzinną hipercholesterolemię, diagnostyką obejmuje się także krewnych. Dokładne określenie mutacji odpowiedzialnej za hipercholesterolemię rodzinną wymaga wykonania badań genetycznych, ale nie wpływa na postępowanie z chorym, czyli sposób leczenia.
Dalsza diagnostyka u pacjenta z rozpoznaną rodzinną hipercholesterolemią ma na celu wykrycie ewentualnych powikłań. Najczęściej przeprowadza się specjalistyczne badania kardiologiczne, angiologiczne i okulistyczne. Do badań oceniających stopień zaawansowania zmian miażdżycowych w naczyniach krwionośnych należą: USG tętnic obwodowych metodą Dopplera (czyli z oceną przepływu krwi), echokardiografia, EKG, próba wysiłkowa, badania angiograficzne (z podaniem kontrastu do ocenianych naczyń), w tym koronarografia oceniająca tętnice wieńcowe oraz nieinwazyjne badania obrazowe naczyń, takie jak angio-TK i angio-MR (tomografia komputerowa i rezonans magnetyczny z podaniem odpowiedniego kontrastu i oceną naczyń krwionośnych). W zależności od sytuacji lekarz specjalista decyduje o wskazaniach do wykonania poszczególnych badań.
Jakie są metody leczenia rodzinnej hipercholesterolemii?
W przeciwieństwie do większości przypadków zwiększonego stężenia cholesterolu, u osób z rodzinną hipercholesterolemią nie można zlikwidować przyczyny zaburzeń. Mutacja genetyczna obecna jest przez całe życie i nieustannie powoduje nieprawidłowości w gospodarce LDL. Nie można zatem wyleczyć przyczyny zwiększonego stężenia LDL, ale można skutecznie je obniżać. Celem leczenia są zatem działania zmniejszające stężenie LDL we krwi. Dzięki temu można uniknąć powikłań, czyli miażdżycy naczyń.
Leczenie zależy od postaci genetycznej choroby – przy nieprawidłowych obu kopiach genu (homozygota), jest to trudna terapia prowadzona w specjalistycznych ośrodkach z personelem, który ma doświadczenie w leczeniu osób z tą chorobą.
Znacznie częstszą heterozygotyczną hipercholestreolemię rodzinną (z jedną nieprawidłową kopią genu) leczy zespół obejmujący lekarza podstawowej opieki zdrowotnej i specjalistów (kardiolog, angiolog, diabetolog, endokrynolog).
Lekarz ustali stężenie cholesterolu LDL, jakie powinno zostać osiągnięte u danego pacjenta z heterozygotyczną hipercholesterolemią rodzinną, aby skutecznie zapobiegać powikłaniom sercowo-naczyniowym. Zależy ono on ryzyka sercowo-naczyniowego.
Sposób leczenia dobiera się w zależności od stężenia cholesterolu LDL we krwi oraz ryzyka sercowo-naczyniowego. Ryzyko sercowo-naczyniowe wylicza się według systemu SCORE. Swoje ryzyko możesz sprawdzić tutaj: Jak oszacować u siebie poziom ryzyka sercowo-naczyniowego?
U osób z małym ryzykiem (<1%) i stężeniem LDL <190 mg/dl lub z umiarkowanym ryzykiem (do <5%) i stężeniem LDL <100 mg/dl leczenie opiera się na zmianie stylu życia. U osób z większym stężeniem cholesterolu LDL i/lub większym ryzykiem sercowo-naczyniowym oprócz zmiany stylu życia wprowadza się leki, czyli farmakoterapię. W zależności od oceny lekarskiej leki wprowadza się po wcześniejszej próbie modyfikacji stylu życia, w razie jej niepowodzenia bądź od razu. W zaawansowanych postaciach czasem stosuje się zabiegi „oczyszczania krwi” z lipoprotein (afereza).
Zmiana stylu życia
W większości przypadków terapię zaczyna się od kilkumiesięcznej próby stosowania się pacjenta do zaleceń dotyczących zmiany stylu życia w celu skutecznego zmniejszenia stężenia LDL; powodzenie takiego postępowania pozwala na długo uniknąć farmakoterapii. Zmniejszenie masy ciała u osób z nadwagą i otyłością przynosi bardzo dobre efekty. Osoby, które nie mają problemów z nadmierną masą ciała, ale chorują na rodzinną hipercholesterolemię, także odnoszą korzyści z poniższych zaleceń.
Pierwszy krok to modyfikacja diety. Ilość przyjmowanych tłuszczów nie powinna przekraczać 30% wszystkich spożywanych kalorii, w tym tłuszczów nasyconych <7%. Należy unikać pokarmów bogatych w tłuszcze nasycone, czyli ograniczyć spożywanie: wołowiny, wieprzowiny, jagnięciny i kurczaków, podrobów i jaj. Zastąpić produkty o pełnej zawartości tłuszczu, produktami niskotłuszczowymi (mleko, nabiał). Tłuszcze nasycone należy zastąpić wielonienasyconymi kwasami tłuszczowymi, które znajdują się w rybach morskich, ziarnach, orzechach, pestkach. Trzeba też unikać spożywania produktów wysoko przetworzonych, gotowych dań, jedzenia typu fast-food, posiłków bogatych w cukry proste i picia kolorowych napojów, w tym soków owocowych. Nowe zasady żywienia warto skonsultować z dietetykiem.
Regularny wysiłek fizyczny zmniejsza skutecznie stężenie cholesterolu we krwi. Wskazane są ćwiczenia przyspieszające rytm serca (tj. bieganie, jazda na rowerze, nordic walking lub pływanie). Co najmniej 30 minut aktywności fizycznej co drugi dzień to dawka sportu działająca leczniczo.
Leki
Jeśli zmiana stylu życia nie przynosi pożądanych efektów lub gdy ryzyko powikłań w postaci choroby układu sercowo-naczyniowego jest duże, należy podjąć leczenie farmakologiczne. Współczesny rynek farmaceutyczny oferuje szeroki wybór leków obniżających stężenie cholesterolu. Ich mechanizm działania bywa różny. Niektóre leki są skuteczniejsze w obniżaniu stężenia LDL, inne triglicerydów, a jeszcze inne zwiększają stężenie HDL („dobrego” cholesterolu).
Lekami najczęściej stosowanymi u chorych na rodzinną hipercholesterolemię zmniejszającymi stężenie LDL są statyny (które także zmniejszają ryzyko zawału serca i udaru), żywice jonowymienne, ezetymib, inhibitory PCSK9, kwas bempediowy. Terapię zaczyna się z reguły od jednego leku, a w razie braku pożądanych efektów terapeutycznych można zastosować terapię skojarzoną kilkoma lekami.
Wiele nowych leków pozostaje w trakcie badań klinicznych.
Afereza
To forma leczenia stosowana najczęściej u osób z dużym stężeniem LDL (>200 mg/dl u osób z chorobą układu sercowo-naczyniowego; >300 mg/dl u osób bez dodatkowego obciążenia) pomimo stosowania innych metod leczenia – najczęściej u chorych na homozygotyczną postać choroby (oba geny z mutacją). Polega na powtarzanych zabiegach usuwania krwi z organizmu, przepuszczaniu jej przez specjalny filtr zatrzymujący cząstki LDL i ponownym wprowadzaniu „oczyszczonej” krwi do organizmu. Zabiegi są zwykle powtarzane co 2 tygodnie. Ponadto ci pacjenci muszą przyjmować leki: statynę w skojarzeniu z inhibitorami PCSK9.
Leczenie powikłań
W sytuacji, kiedy doszło już do zmian miażdżycowych i zwężenia naczyń wieńcowych bądź naczyń obwodowych, niezbędne bywa leczenie mające na celu przywrócenie prawidłowego dopływu krwi do narządów. Poza opisanymi powyżej zasadami leczenia hipercholesterolemii stosuje się dodatkowe leki zalecane przez kardiologa lub angiologa.
Zabiegi kardiologii inwazyjnej – angioplastyka naczyń wieńcowych bądź pomostowanie aortalno-wieńcowe (tzw. bypass), pozwalają przywrócić napływ krwi do mięśnia sercowego w zaawansowanych postaciach choroby niedokrwiennej serca. Zabiegi chirurgii naczyniowej – przezskórne (małoinwazyjne), bądź klasyczne – operacyjne – mają na celu przywrócenie napływu krwi do innych tkanek i narządów.
Istnieją wyspecjalizowane ośrodki posiadające doświadczenie w opiece nad chorymi na rodzinną hipercholesterolemię, które prowadzą specjalne programy lekowe. W Polsce są to następujące placówki:
| Tabela. Wyspecjalizowane ośrodki posiadające doświadczenie w opiece nad chorymi na rodzinną hipercholesterolemię | |||
|---|---|---|---|
| nazwa placówki | adres | województwo | |
| Oddział Kliniczny Kardiologiczny i Poradnia Kardiologiczna Uniwersytecki Szpital Kliniczny Jana Mikulicza-Radeckiego we Wrocławiu | Borowska 213 | 50-556 Wrocław | dolnośląskie |
| Poradnia Kardiologiczna Wojewódzki Szpital Specjalistyczny w Legnicy | Jarosława Iwaszkiewicza 5 59-220 | Legnica | dolnośląskie |
| Poradnia Kardiologiczna Konsultacyjna Wojewódzki Szpital Zespolony im. Ludwika Rydygiera w Toruniu | św. Józefa 53-59 | 87-100 Toruń | kujawsko-pomorskie |
| Poradnia Kardiologiczna Szpital Uniwersytecki nr 2 im. dr. Jana Biziela w Bydgoszczy | Kornela Ujejskiego 75 | 85-168 Bydgoszcz | kujawsko-pomorskie |
| Poradnia Kardiologiczna Samodzielny Publiczny Szpital Kliniczny nr 4 w Lublinie | Kazimierza Jaczewskiego 8 | 20-954 Lublin | lubelskie |
| Poradnia Kardiologiczna Wojewódzki Szpital Specjalistyczny im. Stefana kardynała Wyszyńskiego SP ZOZ | al. Kraśnicka 100 | 20-718 Lublin | lubelskie |
| Poradnia Kardiologiczna Szpital Uniwersytecki im Karola Marcinkowskiego w Zielonej Górze | Zyty 26 | 65-980 Zielona Góra | lubuskie |
| Poradnia Kardiologiczna dla Dorosłych z Wrodzonymi Wadami Serca Instytut Centrum Zdrowia Matki Polki | Rzgowska 281/289 | 93-338 Łódź | łódzkie |
| Poradnia Kardiologiczna SP ZOZ Centralny Szpital Kliniczny Uniwersytetu Medycznego w Łodzi | Pomorska 251 | 92-213 Łódź | łódzkie |
| Poradnia Kardiologiczna Wojewódzki Szpital Specjalistyczny im. dr. Władysława Biegańskiego | gen. Karola Kniaziewicza 1/5 | 91-347 Łódź | łódzkie |
| Poradnia Kardiologiczna Krakowski Szpital Specjalistyczny im. Jana Pawła II | Prądnicka 80 | 31-202 Kraków | małopolskie |
| Poradnia Metabiliczna Szpital Uniwersytecki w Krakowie | Mikołaja Kopernika 15, 15c | 31-501 Kraków | małopolskie |
| Instytut Żywności i Żywienia im. prof. dr. med. Aleksandra Szczygła | Powsińska 61/63 | 02-903 Warszawa | mazowieckie |
| Poradnia Kardiologiczna Samodzielny Publiczny Centralny Szpital Kliniczny | Stefana Banacha 1A | 02-097 Warszawa | mazowieckie |
| Instytut Kardiologii im. Prymasa Tysiąclecia Kardynała Stefana Wyszyńskiego | Alpejska 42 | 04-628 Warszawa | mazowieckie |
| Oddział Kardiologi i Poradnia Kardiologiczna Uniwersytecki Szpital Kliniczny w Opolu | Aleja Wincentego Witosa 26 | 45-401 Opole | opolskie |
| Poradnia Chorób Metabolicznych oraz Poradnia Kardiologiczna Kliniczny Szpital Wojewódzki nr 2 im. św. Jadwigi Królowej w Rzeszowie | Lwowska 60 | 35-301 Rzeszów | podkarpackie |
| Poradnia Kardiologiczna SP ZOZ | Szpitalna 16 | 37-200 Przeworsk | podkarpackie |
| Kliniczne Centrum Kardiologii Uniwersyteckie Centrum Kliniczne | Dębniki 7 | 80-214 Gdańsk | pomorskie |
| Poradnia Kardiologiczna Uniwersyteckie Centrum Kliniczne | Marcina Smoluchowskiego 17 | 80-952 Gdańsk | pomorskie |
| Poradnia Kardiologiczna Dorosłych Śląskie Centrum Chorób Serca w Zabrzu | Marii Skłodowskiej-Curie 9 | 44-100 Zabrze | śląskie |
| Poradnia Kardiologiczna Górnośląskie Centrum Medyczne ŚUM im. prof. Leszka Gieca w Katowicach | Ziołowa 45/47 | 40-634 Katowice | śląskie |
| Poradnia Kardiologiczna Śląskie Centrum Rehabilitacji i Prewencji | Zdrojowa 6 | 43-450 Ustroń | śląskie |
| Poradnia Kardiologii Wojewódzki Szpital Specjalistyczny im. NMP w Częstochowie | Bialska 104/118 | 42-200 Częstochowa | śląskie |
| Poradnia Leczenia Schorzeń Metabolicznych Uniwersyteckie Centrum Kliniczne SUM im. prof. Kornela Gibińskiego w Katowicach | Medyków 14 | 44-001 Katowice | śląskie |
| Poradnia Kardiologiczna Wojewódzki Szpital Zespolony w Kielcach | Grunwaldzka 45 | 25-736 Kielce | świętokrzyskie |
| Poradnia Zaburzeń Metabolicznych Szpital Kliniczny Przemienienia Pańskiego Uniwersytetu Medycznego im. Karola Marcinkowskiego w Poznaniu | Augustyna Szamarzewskiego 82/84 | 60-569 Poznań | wielkopolskie |
| Poradnia Kardiologiczna Wielospecjalistyczny Szpital Miejski im. Józefa Strusia z Zakładem Opiekuńczo-Leczniczym SP ZOZ z siedzibą w Poznaniu | Szwajcarska 3 | 61-285 Poznań | wielkopolskie |
| Poradnia Kardiologiczna oraz Poradnia Chorób Wewnętrzych Samodzielny Publiczny Szpital Kliniczny nr 2 PUM w Szczecinie | al. Powstańców Wielkopolskich 72 | 70-111 Szczecin | zachodniopomorskie |
Czy jest możliwe całkowite wyleczenie rodzinnej hipercholesterolemii?
Z uwagi na genetyczne podłoże zaburzeń, nie jest możliwe całkowite wyleczenie hipercholesterolemii rodzinnej. Można jednak zachować odpowiednią kontrolę i zapobiec powikłaniom choroby. To, w jakim stopniu uda się uniknąć bądź opóźnić wystąpienie chorób układu sercowo-naczyniowego, zależy przede wszystkim od stosowania się pacjenta do zaleceń lekarskich. Zasady dotyczące stylu życia oraz (jeśli to niezbędne) farmakoterapii trzeba stosować do końca życia.
Ryzyko wczesnego zawału serca, udaru oraz przedwczesnej śmierci z przyczyn sercowo-naczyniowych jest zwiększone szczególnie w przypadku hipercholesterolemii homozygotycznej (obie kopie genu z mutacją), gdyż ta postać choroby nie odpowiada tak dobrze na leczenie jak postać heterozygotyczna.
Co trzeba robić po zakończeniu leczenia rodzinnej hipercholesterolemii?
Leczenie hipercholesterolemii rodzinnej musi być bezterminowe, gdyż przez całe życie utrzymują się zaburzenia lipidowe wynikające z mutacji. Pacjenci z rodzinną hipercholesterolemią wymagają regularnych badań kontrolnych z oceną profilu lipidów, a szczególnie stężenia cholesterolu całkowitego i cholesterolu LDL. Wskazana jest także okresowa kontrola specjalistyczna u kardiologa i angiologa.
Co robić, aby uniknąć zachorowania na rodzinną hipercholesterolemię?
Nie da się uniknąć zachorowania na rodzinną hipercholesterolemię, jeśli jest się obciążonym mutacją warunkującą tę chorobę. Można natomiast opóźnić powikłania choroby, stosując się do opisanych powyżej zasad terapeutycznych.