Akromegalia to przewlekła choroba spowodowana nadmiernym wydzielaniem hormonu wzrostu. Jej przyczynę stanowi zwykle łagodny guz (gruczolakorak) przysadki mózgowej. Akromegalia zwykle rozwija się powoli, a do pierwszych objawów należą powiększenie dłoni, stóp i twarzoczaszki, zlewne poty i bóle głowy. W leczeniu stosuje się przede wszystkim operacyjne usunięcie guza przysadki. Akromegalia powoduje wiele powikłań, także potencjalnie zagrażających życiu.
Akromegalia – czym jest i jej przyczyny?
Akromegalia jest chorobą przewlekłą związaną z nadmiernym wydzielaniem hormonu wzrostu (somatotropina). Charakteryzuje się postępującą stopniowo zmianą wyglądu zewnętrznego oraz wieloma powikłaniami narządowymi. Akromegalia występuje u osób dorosłych, u których zakończony został proces wzrastania kości. Natomiast u dzieci i młodzieży nadmiar hormonu wzrostu prowadzi do tzw. gigantyzmu (nadmiernego wzrostu ciała).
Najczęstszą przyczyną nadmiernego wydzielania hormonu wzrostu jest gruczolak (łagodny guz) przysadki. Rzadko (w mniej niż 1% przypadków) nadmierne wydzielanie hormonu wzrostu jest spowodowane pobudzeniem przysadki przez nowotwór neuroendokrynny (wydzielający hormony) oskrzela, przewodu pokarmowego lub trzustki.
Może Cię zainteresować:
- Zespół rakowiaka
- Wybrane nowotwory gruczołów dokrewnych (wydzielania wewnętrznego)
- Rak trzustki - objawy nowotworów złośliwych trzustki - diagnoza i rokowania
Jak działa hormon wzrostu i jego rola w organizmie?
Hormon wzrostu jest wytwarzany przez wyspecjalizowane komórki przysadki, gruczołu położonego w środkowym dole czaszkowym. U dzieci hormon wzrostu jest konieczny dla prawidłowego wzrastania, u dorosłych wpływa na metabolizm w organizmie, czynność kości i mięśni.
Hormon wzrostu prowadzi do wytwarzania przez wątrobę czynników wzrostowych, głównie IGF-1 (insulin-like growth factor-1 – insulinopodobny czynnik wzrostu, inaczej nazywany somatomedyną C). Czynniki wzrostowe są odpowiedzialne za efekty działania hormonu wzrostu.
Jak często występuje akromegalia?
Akromegalia występuje rzadko. Częstość występowania szacuje się na 50–70 osób na milion. Corocznie chorobę stwierdza się u około 4 osób na milion. Ocenia się, że w Polsce na akromegalię choruje ponad 2000 osób. Chorobę obserwuje się dwukrotnie częściej u kobiet.
Akromegalia – objawy
Pierwsze objawy akromegalii
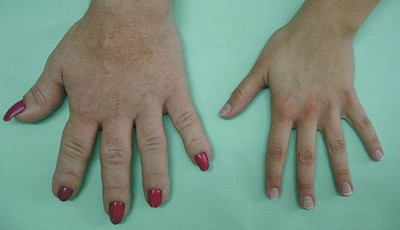
Zmiany wyglądu zewnętrznego typowe dla akromegalii rozwijają się powoli i są przez długi okres niedostrzegalnie dla chorego. Zwykle od pojawienia się pierwszych objawów choroby do rozpoznania akromegalii upływa kilka lat (zwykle 4–10). Dochodzi do powiększenia rąk i stóp, co zmusza chorych w okresie kilku lat do zmiany rozmiaru noszonych butów i rękawiczek na większy (u kobiet pierwszym sygnałem może być konieczność zmiany rozmiaru pierścionków). Powiększeniu ulega również twarzoczaszka – typowy dla akromegalii jest szeroki i gruby nos, powiększenie żuchwy z szerokimi szparami między zębami, wydatne łuki brwiowe. Mogą się pojawić charakterystyczne zlewne poty i częsty ból głowy.
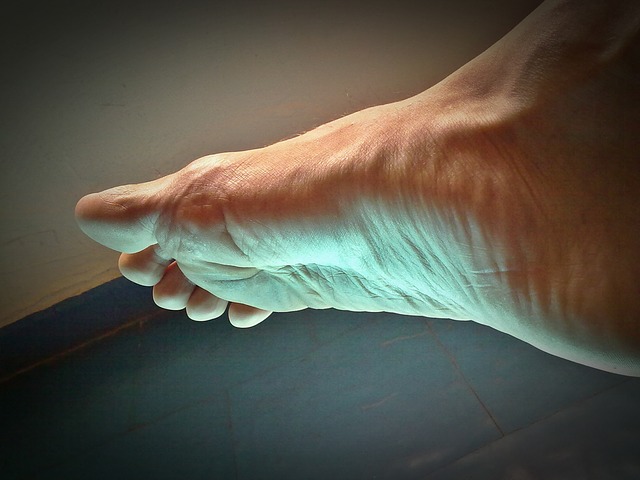
Fot. Powiększenie i obrzmienie ręki - najbardziej charakterystyczny objaw akromegalii
Źródło: Interna Szczeklika, Kraków 2024
Objawy ogólne akromegalii to:
- powiększenie rąk, stóp, twarzoczaszki (nos, żuchwa) i języka, pogrubienie rysów twarzy
- obrzmienie tkanek miękkich (wrażenie opuchnięcia)
- przyrost masy ciała
- wzmożona potliwość
- nadmierne owłosienie
- zmiana barwy głosu.
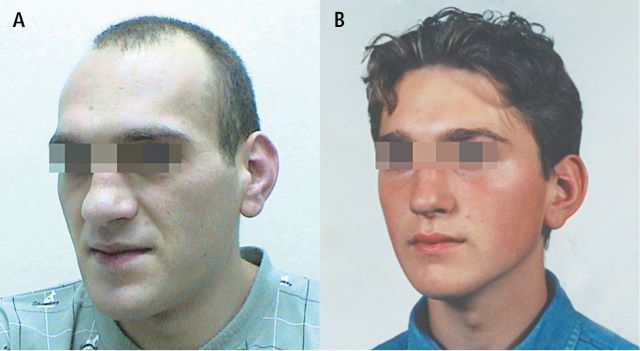
Fot. Objawy akromegalii - zmiany wyglądu twarzoczaszki
Źródło: Interna Szczeklika, Kraków 2024
Objawy akromegalii ze strony poszczególnych układów
Objawy ze strony układu sercowo-naczyniowego
- nadciśnienie tętnicze
- powiększenie serca (tzw. kardiomiopatia przerostowa)
- duszność (niewydolność serca)
- zaburzenia rytmu serca
- w długotrwałej chorobie – wady zastawkowe serca, choroba niedokrwienna serca, udar mózgu.
Objawy ze strony układu oddechowego
- chrapanie, obturacyjny bezdech senny
- upośledzona drożność górnych dróg oddechowych
- w długotrwałej chorobie: rozstrzenie oskrzeli, rozedma płuc.
Zaburzenia metaboliczne i objawy ze strony układu wewnątrzwydzielniczego
- nieprawidłowa tolerancja glukozy lub cukrzyca, hiperinsulinizm (zbyt duże stężenie insuliny)
- hiperlipidemia – zwiększone stężenie cholesterolu i triglicerydów
- hiperkalciuria – zwiększone wydzielanie wapnia z moczem
- wole tarczycy
- nadczynność tarczycy
- mlekotok - wyciek mleka z brodawek sutkowych u kobiet nieciężarnych i niekarmiących, występujący przy ucisku lub samoistnie
- hipogonadyzm – zmniejszone wydzielanie hormonów płciowych
- towarzyszące objawy współistniejącej nadczynności przytarczyc lub guza trzustki.
Objawy ze strony układu pokarmowego
- zaparcie (wydłużenie i poszerzenie jelita grubego)
- ból brzucha, krew w kale (polipy i uchyłki jelita grubego).
Objawy ze strony układu moczowo-płciowego
- zaburzenia miesiączkowania, niepłodność
- mięśniaki macicy
- osłabienie libido
- zaburzenia wzwodu
- łagodny rozrost gruczołu krokowego
- kamica nerkowa.
Objawy ze strony układu nerwowego
- ból głowy
- ograniczenie pola widzenia (przez guza, makrogruczolaka, uciskającego skrzyżowanie nerwów wzrokowych)
- parestezje, niedowłady (m.in. zespół cieśni nadgarstka)
- neuropatie, czyli uszkodzenia nerwów.
Objawy ze strony układu kostno-stawowego
- ból i deformacje kości i stawów
- ograniczona ruchomość stawów
- zmniejszenie gęstości mineralnej kości (osteoporoza).
Co robić w razie wystąpienia objawów akromegalii?
Zwykle na początku pacjent zgłasza się do lekarza rodzinnego, ponieważ objawy akromegalii rozwijają się powoli. Przy podejrzeniu akromegalii lekarz rodzinny kieruje pacjenta do endokrynologa (specjalisty w dziedzinie gruczołów wydzielania wewnętrznego i hormonów), który zleci wykonanie badań potwierdzających rozpoznanie.
Akromegalia – jak lekarz stawia diagnozę?
Przy podejrzeniu akromegalii, lekarz w pierwszej kolejności wykonuje badania krwi obejmujące: ocenę stężenia IGF-1 (u chorych poziom tego czynnika wzrostowego jest podwyższony); test hamowania wydzielania hormonu wzrostu po podaniu glukozy (u osób zdrowych duże stężenia glukozy we krwi hamuje wydzielanie hormonu wzrostu, u chorych z akromegalią obserwuje się brak hamowania).
Po potwierdzeniu rozpoznania lekarz zleca badania obrazowe przysadki, zwykle rezonans magnetyczny w celu uwidocznienia guza i oceny jego wielkości. Zazwyczaj u chorych z akromegalią stwierdza się makrogruczolaki (guzy >1 cm średnicy). W każdym przypadku makrogruczolaka niezbędna jest konsultacja okulisty, który podczas badania oceni dno oka i pole widzenia, ponieważ makrogruczolaki mogą uciskać skrzyżowanie nerwów wzrokowych i powodować tzw. niedowidzenie połowicze dwuskroniowe, czyli ograniczenie pola widzenia od strony skroni (zawężenie pola widzenia). Ponadto w przypadku makrogruczolaka lekarz wykonuje badania krwi w celu wykluczenia niedoczynności przysadki i nadczynności tarczycy.
Jakie są sposoby leczenia akromegalii?
Celem leczenia guzów wydzielających hormon wzrostu jest usunięcie (ewentualnie zmniejszenie) guza oraz zmniejszenie stężenia hormonu wzrostu i IGF-1 do wartości prawidłowych.
Leczenie operacyjne
Najlepszą metodą leczenia jest operacja usunięcia guza przez nos i zatokę klinową, wykonywana w wyspecjalizowanych ośrodkach neurochirurgicznych. Dzięki opracowaniu takiej drogi dojścia do przysadki, zabieg operacyjny wykonuje się bez otwierania czaszki, co zmniejsza ryzyko możliwych powikłań. Takie leczenie może prowadzić do całkowitego wyleczenia, a pacjent nie wymaga dodatkowego leczenia. Niestety całkowite usunięcie dużego guza może być trudne i niekiedy po zabiegu konieczne jest leczenie dodatkowe lub ponowne operacje.
Farmakologiczne leczenie akromegalii
Istnieją leki zmniejszające stężenie hormonu wzrostu i zmniejszające wielkość guza (ale nie prowadzące do jego zniknięcia). Najczęściej stosuje się wstrzyknięcia tzw. analogów somatostatyny I generacji, do których należą lanreotyd, oktreotyd. Leki te można stosować przed planowaną operacją lub po niecałkowitym usunięciu gruczolaka. W razie nieskuteczności analogów somatostatyny I generacji lekarz może zalecić analog somatostatyny II generacji o silniejszym działaniu (pasyreotyd) lub lek o innym działaniu, tzw. antagonistę receptora somatotropinyy (pegwisomant), który skutecznie normalizuje stężenia IGF-1 również u tych pacjentów, u których inne metody leczenia zawiodły. Inną możliwością jest leczenie skojarzone: dołączenie do analogu somatostatyny I generacji pegwisomantu albo tzw. leku dopaminergicznego. Lekarz omawia możliwości leczenia z pacjentem, jego efekty i ewentualne konsekwencje i dobiera najlepszą terapię.
Akromegalia - leczenie uzupełniające (radioterapia)
Jako leczenie uzupełniające w przypadkach nieskutecznego leczenia operacyjnego i farmakologicznego stosuje się radioterapię. Leczenie takie pozwala na zmniejszenie rozmiarów guza i uzyskanie prawidłowych stężeń hormonu wzrostu, ale ostateczny efekt leczenia uzyskuje się dopiero po kilku lub nawet kilkunastu latach, więc do tego czasu konieczne jest leczenie analogami somatostatyny. Zaletą radioterapii jest trwałość uzyskanego efektu leczenia, minusem zaś – poważne działania niepożądane, jak niedoczynność przysadki (nabyty brak wytwarzania jednego lub wielu hormonów przysadki) czy popromienne uszkodzenie nerwu wzrokowego i ślepota (zwykle jednego oka).
Akromegalia – czy możliwe jest całkowite wyleczenie?
Należy podkreślić, że wczesne rozpoznanie akromegalii pozwala na skuteczne leczenie objawów i zapobiega rozwojowi powikłań.
Leczenie operacyjne jest jedyną metodą, która może prowadzić do całkowitego wyleczenia. Leczenie farmakologiczne jest przewlekłe i pozwala jedynie na spowolnienie i kontrolowanie przebiegu choroby.
Akromegalia - rokowanie
Rokowanie co do chirurgicznego wyleczenia akromegalii zależy od wielkości i lokalizacji guza przysadki oraz doświadczenia neurochirurga. Ogólnie skuteczność leczenia waha się od 80% w przypadku mikrogruczolaków do <50% w przypadku guzów o średnicy ponad 1 cm.
W przypadku osób z nieleczoną akromegalią przewidywana długość życia jest średnio krótsza o 10 lat, a śmiertelność z powodu chorób układu sercowo-naczyniowego, oddechowego i chorób nowotworowych 2–4 razy większa niż w populacji ogólnej.
Co trzeba robić po zakończeniu leczenia akromegalii?
Chory wymaga stałej opieki w poradni endokrynologicznej. Po zakończeniu leczenia wskazana jest okresowa kontrola badań krwi i wykonywanie kontrolnych badań obrazowych.
Co robić, aby uniknąć zachorowania na akromegalię?
Sposoby zapobiegania wystąpieniu akromegalii nie są obecnie znane.
Akromegalia - pytania i odpowiedzi
Czy akromegalia jest dziedziczna?
Nie, akromegalia nie jest chorobą dziedziczną, ale może się kojarzyć z pierwotną nadczynnością przytarczyc i nowotworami neuroendokrynnymi trzustki w zespole MEN1 oraz zespołem Carneya i zespołem McCune’a i Albrighta.
Czy akromegalia powoduje cukrzycę?
Tak, w przebiegu akromegalii może dojść do rozwoju cukrzycy.
Czy akromegalia zawsze powoduje powiększenie rąk, stóp i twarzy?
Powiększenie rąk, stóp i twarzy jest jednym z pierwszych objawów akromegalii.
Czy akromegalia to choroba śmiertelna?
Tak, nieleczona akromegalia może prowadzić do wielu powikłań i zgonu. W przypadku osób z nieleczoną akromegalią przewidywana długość życia jest krótsza średnio o 10 lat, a śmiertelność z powodu chorób układu sercowo-naczyniowego, oddechowego i chorób nowotworowych 2–4 razy większa niż w populacji ogólnej.
Czy akromegalia wpływa na płodność?
Tak, akromegalia może powodować zaburzenia miesiączkowania i niepłodność u kobiet, a także zaburzenia wzwodu u mężczyzn.
Czy akromegalia zawsze skraca życie?
Trudno odpowiedzieć na takie pytanie. Wiadomo, że nieleczona akromegalia prowadzi do wielu powikłań, w tym potencjalnie zagrażających życiu, a osoby z nieleczoną akromegalią żyją średnio 10 lat krócej niż populacja ogólna.