
Postać o późnym początku
Poradnictwo genetyczne
Choroba Pompego jest dziedziczona autosomalnie
recesywnie. Oznacza to, że rodzice chorego dziecka
ponoszą 25% ryzyko powtórzenia się tej samej choroby u kolejnych dzieci. Ryzyko to dotyczy każdej
ciąży i nie zależy od płci dziecka. Przebieg kliniczny
choroby będzie podobny u wszystkich chorych
dzieci tych samych rodziców (tzn. jeżeli u jednego
dziecka wystąpiła ciężka postać o wczesnym
początku, kolejne chore dziecko również będzie
miało taką postać choroby).
Jeżeli znane są mutacje patogenne u chorego
dziecka, w kolejnej ciąży można rozważyć przeprowadzenie
diagnostyki prenatalnej w kierunku
choroby Pompego. Podejmując decyzje prokreacyjne,
rodzice mogą również rozważyć diagnostykę przedimplantacyjną
(możliwą do wykonania w połączeniu z procedurą zapłodnienia pozaustrojowego).
Ryzyko wystąpienia choroby u dziecka chorej
osoby jest niewielkie, ale każde dziecko takiej osoby
będzie nosicielem mutacji patogennej w jednym
allelu. W tej sytuacji choroba u dziecka mogłaby
wystąpić tylko w przypadku, gdyby drugi z rodziców
(tzn. partner lub partnerka chorej osoby) był
nosicielem mutacji patogennej tego samego genu.
Biorąc pod uwagę rzadkie występowanie choroby
Pompego, taki stan nosicielstwa u partnera/partnerki
jest mało prawdopodobny, ale nie można go
wykluczyć bez wykonania badań genetycznych.
Należy również pamiętać, że prawdopodobieństwo
nosicielstwa mutacji może być znacznie większe,
jeżeli partner/partnerka wykazuje pokrewieństwo z pacjentem. Opisano przypadki urodzenia
dziecka przez kobiety otrzymujące enzymatyczną
terapię zastępczą
W każdym przypadku rozpoznania choroby
Pompego wskazana jest konsultacja rodziny w poradni genetycznej.
Leczenie
Postępowanie terapeutyczne obejmuje leczenie przyczynowe za pomocą enzymatycznej terapii zastępczej oraz leczenie objawowe poszczególnych zaburzeń, np. niewydolności serca lub niewydolności oddechowej.
Enzymatyczna terapia zastępcza
Leczenie przyczynowe polega na przewlekłej
substytucji alglukozydazy alfa, rekombinowanego
analogu naturalnego enzymu. Lek podaje
się w powolnym wlewie dożylnym co dwa tygodnie w dawce 20 mg/kg. W Polsce terapia jest w całości refundowana, zarówno u dzieci, jak i dorosłych bez ograniczenia wieku, w ramach
programu leczenia chorób ultrarzadkich. Szpital, w którym będzie prowadzone leczenie, musi złożyć
odpowiedni wniosek do Zespołu Koordynacyjnego
ds. Chorób Ultrarzadkich. Warunkiem kwalifikacji
pacjenta do leczenia jest potwierdzenie rozpoznania
zarówno za pomocą testów enzymatycznych,
jak i genetycznych. Zgodnie z wymogami
programu terapeutycznego przed rozpoczęciem
leczenia i następnie co 6 miesięcy należy przeprowadzać
dokładną ocenę stanu klinicznego
pacjenta, m.in. z uwzględnieniem echokardiografii,
spirometrii oraz testów sprawności ruchowej. W razie nieskuteczności leczenia zgoda na refundację
terapii może zostać wstrzymana.
Ze względu na ryzyko reakcji nadwrażliwości, w tym wstrząsu anafilaktycznego, oraz ogólnie
zwiększone ryzyko powikłań z powodu niewydolności
oddechowej i/lub niewydolności serca
leczenie należy prowadzić w szpitalu dysponującym
odpowiednim oddziałem intensywnej opieki
medycznej.
Ryzyko reakcji nadwrażliwości jest większe u niemowląt całkowicie pozbawionych naturalnego
enzymu, u których wynik testu Western blot
na obecność alfa-glukozydazy w fibroblastach
skóry jest ujemny. Większe jest również u nich
ryzyko powstania przeciwciał znacząco hamujących
działanie egzogennego enzymu. Z tego
powodu w czasie enzymatycznej terapii zastępczej u tych dzieci proponuje się równoczesne
stosowanie leków immunomodujących, najczęściej
rytuksymabu w połączeniu z innym lekiem
immunosupresyjnym.
Wyniki dotychczasowych badań klinicznych
wskazują, że u niemowląt i młodszych dzieci
(<3,5 lat) enzymatyczna terapia zastępcza alglukozydazą
alfa wydłuża przeżycie i okres bez
konieczności stosowania wentylacji mechanicznej
oraz zmniejsza masę serca i znacząco poprawia rozwój ruchowy. Skuteczność leczenia jest na ogół
tym większa, im wcześniej rozpoczęto leczenie.
U pacjentów z postacią o późnym początku leczenie
poprawia sprawność ruchową i zatrzymuje
progresję niewydolności oddechowej.
Odległe efekty leczenia są trudne do oceny
ze względu na małą liczbę pacjentów oraz krótki
jak dotąd okres obserwacji. Poszczególne typy
mięśni mogą być w różnym stopniu podatne
na leczenie, ale nie ustalono dokładnie, jakie
inne czynniki poza przeciwciałami przeciw alglukozydazie
alfa wpływają na skuteczność terapii.
Dostępne obecnie dane naukowe świadczą jednak
zgodnie, że większość leczonych odnosi istotne
korzyści kliniczne.
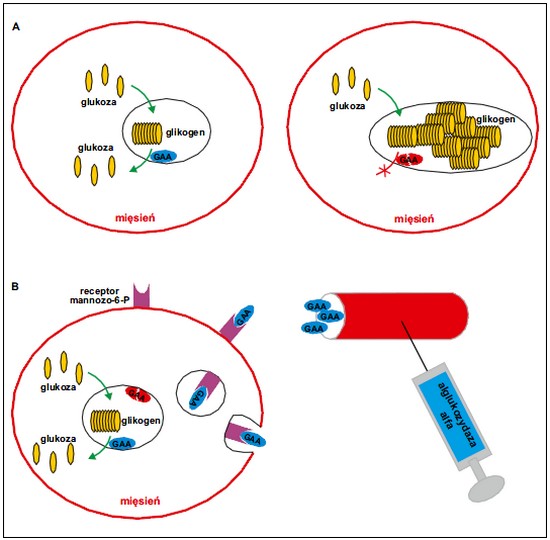
Ryc. 1. Patomechanizm choroby Pompego i zasada działania enzymatycznej terapii zastępczej. W następstwie wrodzonego niedoboru alfa-glukozydazy (GAA) w lizosomach mięśni szkieletowych i mięśnia sercowego gromadzi się w nadmiarze glikogen (A). Leczenie polega na okresowym podawaniu alglukozydazy alfa (syntetycznego analogu alfa-glukozydazy), która za pośrednictwem receptorów mannozo-6-fosforanu przenoszona jest do lizosomów, kompensując wrodzony defekt genetyczny (B)
Leczenie objawowe
Poszczególne objawy choroby, takie jak niewydolność
serca lub niewydolność oddechowa, wymagają
zazwyczaj standardowego postępowania zalecanego w tych sytuacjach niezależnie od przyczyny. U dzieci z kardiomiopatią przerostową w leczeniu
niewydolności serca przeciwwskazane może być
stosowanie digoksyny i innych leków nasilających
zwężenie drogi odpływu z lewej komory. Wskazane
jest energiczne zwalczanie zakażeń dróg oddechowych,
przestrzeganie programu szczepień
ochronnych i stosowanie odpowiedniej rehabilitacji
oddechowej. W celu zapobiegania zakażeniom
syncytialnym wirusem oddechowym w pierwszych 2 latach życia zaleca się stosowanie paliwizumabu. W miarę możliwości należy unikać znieczulenia
ogólnego, zwłaszcza u najmłodszych dzieci z przerostem
mięśnia sercowego, ale także u pacjentów z chorobą Pompego o późniejszym początku.
Konieczne jest regularne monitorowanie stanu
odżywienia oraz prowadzenie rehabilitacji ruchowej i fizjoterapii odpowiednio do stanu klinicznego i wieku chorego.
Najnowsze dane wskazują na zwiększone
ryzyko poszerzenia aorty i tętniaków wewnątrzczaszkowych u chorych na postać o późnym
początku, co należy uwzględnić w planowaniu
badań profilaktycznych.
Patomechanizm
Przyczyną choroby Pompego jest niedobór alfa-glukozydazy,
hydrolazy lizosomalnej, która uczestniczy w procesie rozkładu glikogenu do glukozy. W efekcie glikogen odkłada się w nadmiarze w komórkach różnych tkanek, zwłaszcza mięśnia
sercowego i mięśni szkieletowych, w tym mięśni
oddechowych (ryc. 1A), powodując przerost i niewydolność
serca (w przypadku skrajnego niedoboru),
niedowład wiotki oraz niewydolność oddechową.
Enzymatyczna terapia zastępcza polega
na dostarczeniu syntetycznego analogu brakującego
enzymu drogą dożylną. Cząsteczki enzymu
zawierają reszty mannozo-6-fosforanu, które
są rozpoznawane przez odpowiednie receptory
na powierzchni komórek, dzięki czemu syntetyczny
enzym przenoszony jest bezpośrednio
do lizosomów we wnętrzu komórki (ryc. 1B).
Najważniejsze wnioski
– W zależności od stopnia niedoboru enzymu choroba Pompego może ujawnić się w każdym okresie życia. U większości chorych występuje postać o późnym początku, przebiegająca zwykle pod postacią nieswoistej miopatii, przypominającej dystrofię obręczowo-kończynową, zazwyczaj bez przerostu mięśnia sercowego.– Chorobę można łatwo wykluczyć za pomocą prostego testu przesiewowego.
– Dostępne jest przyczynowe leczenie choroby za pomocą enzymatycznej terapii zastępczej.
– W Polsce leczonych jest obecnie około 30 pacjentów, podczas gdy chorych może być nawet 1000 osób (według szacunków opartych na danych epidemiologicznych z innych krajów europejskich).
Dane kontaktowe
Aktualne informacje na temat możliwości wykonania enzymatycznych badań przesiewowych, badań genetycznych oraz uzyskania poradnictwa genetycznego dla chorych na chorobę Pompego można uzyskać u autora artykułu:dr med. Marek Bodzioch
Zakład Neurogenetyki, Uniwersytet Jagielloński Collegium Medicum
ul. Botaniczna 3, 31-503 Kraków
tel. 12 424 86 15
e-mail: mbodzioch@cm-uj.krakow.pl















