Skróty: CHMP – Komitet ds. Produktów Leczniczych Stosowanych u Ludzi, EMA – Europejska Agencja Leków, KE – Komisja Europejska, NDL – niepożądane działanie leku, NOP – niepożądany odczyn poszczepienny, UE – Unia Europejska, URPL – Urząd Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Wprowadzenie
Szczepionki, tak jak pozostałe produkty lecznicze, przed wprowadzeniem na rynek wymagają uzyskania pozwolenia na dopuszczenie do obrotu. Wymagania dotyczące ich rejestracji są jednak bardzo rygorystyczne, ponieważ należą one do najbardziej złożonej grupy produktów, czyli biologicznych i ich stosowanie wiąże się z pewnym ryzykiem. W Polsce dopuszczeniem szczepionek do obrotu zajmuje się Prezes Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych (URPL), który podejmuje decyzje na podstawie wniosków podmiotów odpowiedzialnych i załączonej do nich dokumentacji (określonych w art. 10 ustawy Prawo farmaceutyczne), bądź przez Radę Unii Europejskiej (UE) lub Komisję Europejską (KE) po przeprowadzeniu przez Europejską Agencję Leków (European Medicines Agency – EMA) procedury scentralizowanej. W każdym przypadku pozwolenie na dopuszczenie do obrotu jest wydawane po ocenie stosunku korzyści do ryzyka na podstawie dokumentacji zawierającej dane zebrane w czasie prac rozwojowych nad danym produktem i badań klinicznych. Ocena ryzyka dotyczy jakości, bezpieczeństwa i skuteczności produktu leczniczego. Przed wydaniem pozwolenia właściwe organy sprawdzają zgodność danych zawartych w przedstawionej dokumentacji z dobrą praktyką wytwarzania (Good Manufacturing Practice – GMP), dobrą praktyką badań klinicznych (Good Clinical Practice – GCP) i dobrą praktyką laboratoryjną (Good Laboratory Practice – GLP).
Rodzaje procedur rejestracyjnych i dopuszczenie do obrotu
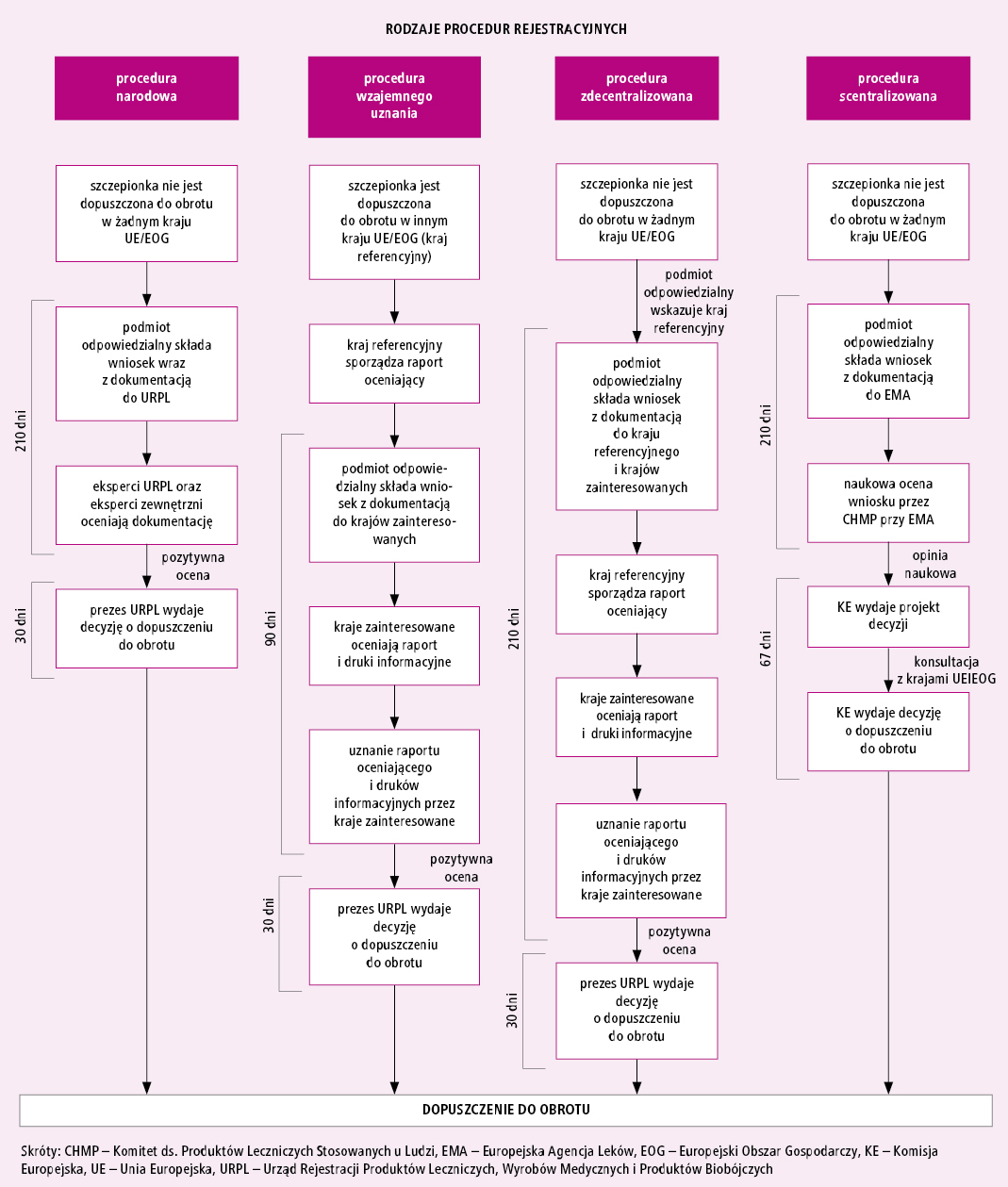
Poza pewnymi ograniczeniami firmy farmaceutyczne mogą dowolnie wybierać rodzaj procedury rejestracyjnej. Obecnie w rejestracji szczepionek najczęściej stosuje się procedurę wzajemnego uznania, procedurę zdecentralizowaną oraz scentralizowaną. Wnioskodawcy najrzadziej wybierają procedurę narodową, zwykle korzystając z niej w przypadku rozszerzenia asortymentu (np. rejestracja nowej postaci farmaceutycznej lub drogi podania).
Procedura narodowa
W procedurze narodowej (National Procedure – NP) szczepionki są dopuszczane do obrotu na podstawie przepisów ustawy Prawo farmaceutyczne oraz rozporządzeń wykonawczych do tej ustawy. W celu zarejestrowania szczepionki konieczne jest złożenie wniosku z wymaganymi załącznikami i dokumentacją dotyczącą jakości, bezpieczeństwa i skuteczności danego preparatu. Po złożeniu dossier rejestracyjnego sprawdzane jest, czy wniosek wypełniono prawidłowo i czy dołączono wszystkie wymagane dokumenty. Dokumentację ocenia zespół ekspertów URPL we współpracy z ekspertami zewnętrznymi, specjalistami z danej dziedziny. W przypadku pozytywnego wyniku postępowania prezes URPL wydaje pozwolenie na dopuszczenie do obrotu produktu leczniczego. Pozwolenie wydane przez prezesa URPL w procedurze narodowej jest ważne tylko na terenie Polski. Procedura ta ma pewne ograniczenia – nie ma zastosowania, jeśli szczepionka należy do produktów biotechnologicznych lub jest szczepionką pandemiczną, a także gdy jest już zarejestrowana w innym kraju członkowskim. Proces dopuszczenia do obrotu w procedurze narodowej trwa 210 dni (p. ryc. 1.).
Ryc. 1. Rodzaje procedur rejestracyjnych produktów leczniczych.
Procedura wzajemnego uznania
Procedurę wzajemnego uznania (Mutual Recognition Procedure – MRP) stosuje się w celu uzyskania pozwolenia na dopuszczenie do obrotu w kilku krajach członkowskich jednocześnie, kiedy dana szczepionka w chwili złożenia wniosku posiada już pozwolenie na dopuszczenie do obrotu w jednym z krajów członkowskich. Jest to procedura dwuetapowa, która obejmuje:
- ocenę dossier i sporządzenie raportu oceniającego przez kraj referencyjny (Reference Member State – RMS)
- uznanie raportu przygotowanego przez RMS przez kraje zainteresowane (Concerned Member State – CMS).
Zainteresowane kraje oceniają dany produkt leczniczy na podstawie raportu przygotowanego przez kraj referencyjny. Procedura wzajemnego uznania trwa 90 dni. Po jej zakończeniu kraje zainteresowane mają 30 dni na weryfikację złożonych przez podmiot odpowiedzialny narodowych wersji druków informacyjnych oraz wydanie narodowej decyzji o dopuszczeniu do obrotu. W Polsce pozwolenie na dopuszczenie do obrotu produktu leczniczego wydaje prezes URPL (p. ryc. 1.).
Procedura zdecentralizowana
Procedura zdecentralizowana (Decentralized Procedure – DCP) jest alternatywą dla procedury wzajemnego uznania. Stosuje się ją w celu uzyskania pozwolenia na dopuszczenie do obrotu w kilku krajach członkowskich jednocześnie, w przypadku gdy dany produkt leczniczy jeszcze nie uzyskał pozwolenia na dopuszczenie do obrotu w żadnym innym kraju członkowskim w chwili złożenia wniosku. Podmiot odpowiedzialny wskazuje kraj referencyjny, który ma za zadanie sporządzenie raportu oceniającego. Kraje zainteresowane wydają swoją decyzję na podstawie raportu oceniającego przygotowanego przez kraj referencyjny.Procedura zdecentralizowana trwa 210 dni. Po jej zakończeniu kraje, które w niej uczestniczą (referencyjny oraz zainteresowane), mają 30 dni na weryfikację złożonych przed podmiot odpowiedzialny krajowych wersji druków informacyjnych oraz wydanie krajowej decyzji o dopuszczeniu do obrotu. W Polsce decyzję o dopuszczeniu do obrotu również wydaje Prezes URPL (p. ryc. 1.).
Procedura scentralizowana
W procedurze scentralizowanej (Centralized Procedure – CP) wniosek o dopuszczenie do obrotu rozpatruje EMA. Pod względem naukowym ocenia go działający w ramach EMA Komitet ds. Produktów Leczniczych Stosowanych u Ludzi (Committee for Medicinal Products for Human Use – CHMP), który wydaje opinię naukową. Przygotowana opinia trafia do KE, która przygotowuje projekt decyzji. Po konsultacji z krajami członkowskimi KE wydaje decyzję o przyznaniu pozwolenia na dopuszczenie do obrotu, które jest ważne w całej UE oraz krajach Europejskiego Stowarzyszenia Wolnego Handlu (European Free Trade Association – EFTA), tj. Islandii, Norwegii i Lichtensteinie (p. ryc. 1.). Ta procedura jest obowiązkowa w przypadku niektórych produktów, takich jak szczepionki wytwarzane z zastosowaniem procesów biotechnologicznych oraz produktów o ogólnym znaczeniu dla zdrowia publicznego w UE, takich jak szczepionki pandemiczne.
Wymagania i ocena dokumentacji składanej z wnioskiem o dopuszczenie do obrotu
Wymagania dotyczące dokumentacji
Niezależnie od rodzaju procedury rejestracyjnej
zakres dokumentacji dołączanej wraz z wnioskiem o dopuszczenie do obrotu szczepionki określony
jest w załączniku 1. do dyrektywy numer 2001/83/WE Parlamentu Europejskiego i Rady z dnia 6 listopada
2001 roku w sprawie wspólnotowego kodeksu
odnoszącego się do produktów leczniczych
stosowanych u ludzi oraz w naukowych wytycznych
przygotowanych i opublikowanych przez
EMA. Szczepionki rejestrowane są jako produkty
oryginalne, nie mogą być produktami generycznymi,
dlatego dokumenty dołączone do wniosku
muszą zawierać wyniki, streszczenia oraz sprawozdania z badań farmaceutycznych, fizykochemicznych,
biologicznych lub mikrobiologicznych,
nieklinicznych: farmakologicznych i toksykologicznych,
oraz klinicznych wraz z ogólnym podsumowaniem
jakości, przeglądem nieklinicznym i streszczeniem danych nieklinicznych oraz przeglądem
klinicznym i podsumowaniem klinicznym.
Cechą odróżniającą tę grupę produktów (biologicznych)
od leków chemicznych jest sposób udokumentowania
jakości substancji czynnej. W każdym
przypadku wnioskodawca zobowiązany jest
do przedstawienia pełnej dokumentacji z zakresu
wytwarzania substancji czynnej (Moduł 3.2.S.),
dokładnie opisującej materiały wyjściowe, proces
wytwarzania substancji czynnej oraz metody i wyniki
badań analitycznych.
Wymagania dotyczące przygotowywania dokumentacja
są bardzo restrykcyjne. Szczepionki,
oprócz wymagań określonych dla wszystkich
produktów leczniczych, muszą spełniać także
dodatkowe warunki określone tylko dla tej grupy
produktów. EMA przygotowała wytyczne
dotyczące szczepionek – scientific guidelines on
vaccines (p. www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/multidisciplinary/multidisciplinary-vaccines) – które powinni uwzględniać producenci. Zakres
wytycznych jest bardzo szeroki: od adiuwantów,
poprzez badania kliniczne, druki informacyjne,
po bezpieczeństwo. Obowiązujące wymagania
dla szczepionek stosowanych u ludzi określone
są również w monografiach (ogólnej i szczegółowych)
Farmakopei Europejskiej/Farmakopei
Polskiej, które określają szczegółowy proces wytwarzania,
etapy jego kontroli, zakres kontroli
oraz metody badań. Kontrola dotyczy nie tylko
substancji czynnej, ale wszystkich materiałów
stosowanych w procesie wytwarzania. Monografie
określają również odczynniki, które można
stosować w procesie wytwarzania, a także substancje
pomocnicze zawarte w szczepionce, dla
których Farmakopea przedstawia rygorystyczne
limity, co gwarantuje bezpieczeństwo stosowania
produktu przez pacjenta. Ponadto przestrzeganie
tych wymagań pozwala zapewnić otrzymywanie
powtarzalnych serii o udowodnionej skuteczności,
immunogenności i bezpieczeństwie dla człowieka.
Obowiązkowym elementem dokumentacji rejestracyjnej
szczepionki są wyniki badań klinicznych
potwierdzające skuteczność i bezpieczeństwo
danego preparatu. Wymagania dotyczące badań
skuteczności i bezpieczeństwa szczepionek są
ostrzejsze niż w odniesieniu do innych produktów
leczniczych. Poszczególne fazy badań klinicznych
wymagają większej liczby uczestników. Wszystkie
badania kliniczne podlegają restrykcyjnym
zasadom etycznym i wymaganiom GCP. Badania
należy prowadzić według wytycznych komitetu naukowego
CHMP Europejskiej Agencji Leków, które
są powiązane z wytycznymi Międzynarodowej
Konferencji ds. Harmonizacji (ICH) i Światowej
Organizacji Zdrowia (WHO). Oprócz szczegółowego
sprawozdania z przeprowadzonych badań klinicznych
przedstawiany jest również plan badań
skuteczności i bezpieczeństwa po wprowadzeniu
produktu na rynek.
Ocena dokumentacji
Każdy dopuszczony do obrotu produkt leczniczy musi się charakteryzować wysoką jakością, bezpieczeństwem stosowania i skutecznością, które są wnikliwie oceniane w trakcie procesu rejestracji. Ocena dokumentacji dołączonej do wniosku o dopuszczenie do obrotu szczepionki odbywa się głównie na podstawie danych naukowych i wytycznych EMA oraz zawartych w „Farmakopei Europejskiej”. Dokumentację ocenia doświadczony zespół ekspertów. Jako wsparcie naukowe przy CHMP działa szereg grup roboczych, m.in. Vaccines Working Party oraz Biological Working Party, do których należy przygotowanie i aktualizacja wytycznych, wspieranie oceny dokumentacji i udzielanie porad naukowych. Jeśli eksperci oceniający dokumentację stwierdzą braki lub nieprawidłowości w dokumentacji rejestracyjnej w trakcie dopuszczenia do obrotu, wnioskodawca jest zobowiązany do rozwiązania problemu i złożenia wymaganych uzupełnień i wyjaśnień w terminach obowiązujących w danej procedurze. Tylko szczepionki w pełni zgodne z przepisami obowiązującymi w UE dotyczącymi jakości, bezpieczeństwa i skuteczności mogą być dopuszczone do obrotu w Polsce.
Rola URPL w nadzorze nad bezpieczeństwem stosowania szczepionek
Monitorowanie działań niepożądanych
Szczepionki to produkty lecznicze, które muszą
spełniać najwyższe standardy bezpieczeństwa.
Jest to zrozumiałe, ponieważ podaje się je zdrowym
osobom, najczęściej dzieciom. Osoba zaszczepiona
może nigdy nie mieć kontaktu z czynnikiem
zakaźnym, przeciwko któremu jest szczepiona. W takiej sytuacji korzyści ze szczepienia odnosi
nie jednostka, ale populacja, a ewentualne ryzyko
ponosi tylko osoba zaszczepiona.
Szczepionki, jak wszystkie grupy leków, podlegają
ciągłemu nadzorowi i ocenie. W tym celu zbiera
się opisy przypadków reakcji niepożądanych i analizuje, czy korzyści ze szczepienia przeważają
nad możliwym do przewidzenia ryzykiem. Kwestię
zgłaszania reakcji poszczepiennych reguluje ustawa
Prawo farmaceutyczne oraz ustawa o zapobieganiu
oraz zwalczaniu zakażeń i chorób zakaźnych u ludzi i rozporządzenie wykonawcze do tej ustawy,
czyli rozporządzenie Ministra Zdrowia w sprawie
niepożądanych odczynów poszczepiennych oraz
kryteriów ich rozpoznawania. W związku z tym
URPL otrzymuje opisy przypadków reakcji poszczepiennych
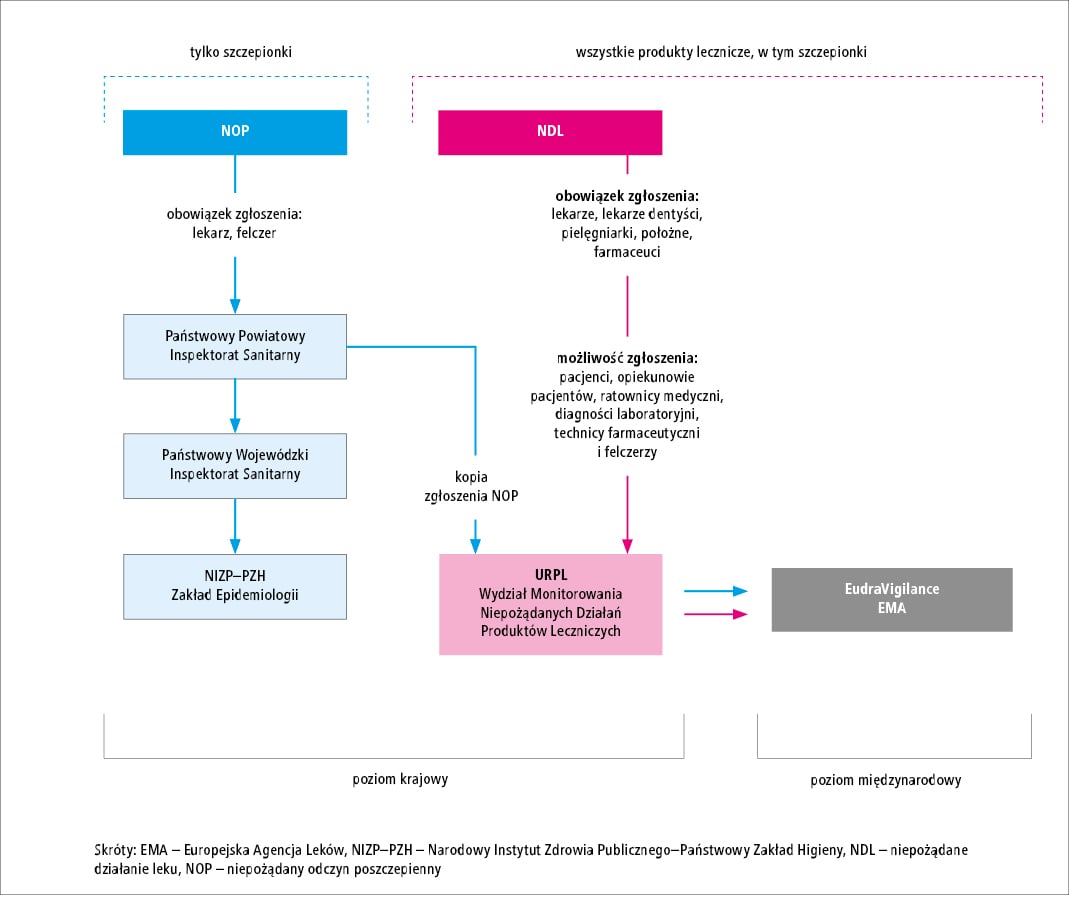
dwiema drogami (p. ryc. 2.).
Ryc. 2. Miejsce URPL w monitorowaniu bezpieczeństwa szczepień.
Pierwsza i zasadnicza droga wynika z realizacji
zobowiązań regulowanych przez wspomniane
rozporządzenie. Głównym adresatem tych zgłoszeń
jest Narodowy Instytut Zdrowia Publicznego–Państwowy
Zakład Higieny (NIZP–PZH), a URPL
otrzymuje ich kopie. System ten polega na tym, że
lekarz lub felczer ma obowiązek wysyłania zgłoszenia
niepożądanego odczynu poszczepiennego (NOP)
na odpowiednim formularzu (który stanowi załącznik
do Rozporządzenia) do państwowego powiatowego
inspektora sanitarnego. Formularz po kolejnym
uzupełnieniu danych trafia do państwowego
wojewódzkiego inspektora sanitarnego. Docelowo
wszystkie zgłoszenia trafiają do NIZP–PZH, gdzie
są oceniane i analizowane. URPL po otrzymaniu
kopii tych zgłoszeń ocenia związek przyczynowo-skutkowy
między podaniem szczepionki a reakcją,
która wystąpiła po szczepieniu (p. ryc. 2.).
Drugą drogę określają przepisy ustawy Prawo
farmaceutyczne. Działania niepożądane związane
ze szczepieniem zgłaszane są według tych samych
reguł, które obowiązują w przypadku pozostałych
produktów leczniczych. Obowiązek prawny przekazywania
opisów przypadków spoczywa na lekarzach,
lekarzach dentystach, pielęgniarkach,
położnych, farmaceutach, ale możliwość taką
mają również pacjenci i ich opiekunowie, ratownicy
medyczni, diagności laboratoryjni, technicy
farmaceutyczni i felczerzy. Osoby zobowiązane
lub uprawnione do przekazania opisu przypadku
mogą wybrać adresata takiego zgłoszenia. Adresatem
może być URPL lub podmiot odpowiedzialny,
czyli firma farmaceutyczna, która jest właścicielem
szczepionki.
URPL ocenia opisy przypadków, które otrzymał
jedną z opisanych dróg, a następnie tłumaczy je
na język angielski i przesyła do europejskiej bazy
danych EudraVigilance (p. ryc. 2.). Taki sam obowiązek
spoczywa na podmiotach odpowiedzialnych
(firmach farmaceutycznych) w odniesieniu
do swoich produktów (jeżeli zostaną wybrane jako
adresat informacji o działaniu niepożądanym).
Niepożądane działanie leku a niepożądany odczyn poszczepienny
Procedury wyglądają dosyć zawile, ale sprawa jest
skomplikowana nie tylko od strony formalno-prawnej,
ale także od strony merytorycznej. Wynika to
przede wszystkim z tego, co rozumiemy pod pojęciem
niepożądanego działania leku (NDL), a co
pod pojęciem NOP. Przytaczając definicję, NDL
to każde niekorzystne i niezamierzone działanie
tego produktu (niezależnie od tego, czy jest on podawany w prawidłowy czy nieprawidłowy sposób).
Jedynym warunkiem jest właściwa jakość produktu (czyli w tym przypadku szczepionka musi być
dobrej jakości). Natomiast NOP jest to zaburzenie
stanu zdrowia czasowo związane ze szczepieniem,
które może być wynikiem: indywidualnej reakcji
organizmu człowieka szczepionego na działanie
szczepionki, błędu wykonania szczepionki lub
błędu podania szczepionki, a także zjawisk niezależnych
od szczepienia, a tylko przypadkowo pojawiających
się po szczepieniu. Czas, jaki bierzemy
pod uwagę w odniesieniu do wszystkich szczepionek,
wynosi 4 tygodnie, z wyjątkiem szczepionki
przeciwko gruźlicy (BCG), dla której ten okres
przedłużony jest do roku. Porównując te definicje, w przypadku NDL mówimy o lekach dobrej jakości, w przypadku NOP mówimy o szczepionkach dobrej
lub złej jakości. Z tego powodu czas na przesłanie
informacji o NOP jest dużo krótszy, bo teoretycznie
może dotyczyć sytuacji, w której zachodzi konieczność
pilnego wycofania konkretnej serii produktu z rynku. Druga różnica to taka, że w odniesieniu
do NDL rozpatrujemy przypadki, w których przynajmniej
nie można wykluczyć związku przyczynowo-skutkowego
między szczepieniem a reakcją u pacjenta. Zakłada się, że osoba zgłaszająca nie
podejmowałaby trudu przygotowania opisu przypadku,
gdyby była przeświadczona, że takiego
związku nie ma. Natomiast w przypadku NOP
należy zgłaszać także przypadki, gdy może nie być
związku przyczynowo-skutkowego, a jest jedynie
związek czasowy. Trzeba pamiętać, że oprócz NOP
istnieje też odczyn poszczepienny, czyli fizjologiczna
reakcja organizmu na szczepienie. Od oceny
osoby badającej pacjenta zależy, jakie reakcje uzna
za odczyn poszczepienny, a jakie za NOP.
URPL, jako jednostka zajmująca się NDL, w tym przede wszystkim wykrywaniem nowych
zagrożeń związanych z farmakoterapią (przykładem
nowego działania niepożądanego wykrytego
swego czasu w Szwecji była możliwość wystąpienia
narkolepsji po podaniu szczepionki przeciwko
grypie pandemicznej), skupia się na przypadkach, w których istnieje związek przyczynowo-skutkowy
między szczepieniem a reakcją.
Zbieranie i analiza opisów pojedynczych przypadków
NDL należy do rutynowych metod minimalizacji
ryzyka stosowania produktów leczniczych.
Na podstawie oceny danych uaktualnia się
informację o leku, wprowadzając – o ile zachodzi
taka potrzeba – ograniczenia jego stosowania.
Dodatkowe działania w zakresie bezpieczeństwa
Do działań pozarutynowych należy m.in. dodatkowe
monitorowanie leków, prowadzenie porejestracyjnych
badań bezpieczeństwa lub skuteczności,
wydawanie komunikatów bezpieczeństwa
oraz materiałów edukacyjnych. Dodatkowe
monitorowanie polega na wybraniu leków podlegających
szczególnie intensywnemu zbieraniu
danych. Do leków tych zalicza się nowe leki (w rozumieniu
nowej cząsteczki), ale także wszystkie
leki biologiczne, a więc także szczepionki. Ulotki
tych leków oraz Charakterystyki Produktów
Leczniczych oznaczone są symbolem czarnego
odwróconego trójkąta i zawierają apel o zgłaszanie
NDL. W ramach porejestracyjnych badań
bezpieczeństwa ocenia się ryzyko towarzyszące
terapii lub sprawdza się sposób stosowania danego
leku w codziennej praktyce klinicznej, a w niektórych
przypadkach ocenia się, czy zaproponowane w przeszłości narzędzia ograniczenia ryzyka
przyniosły oczekiwany skutek. Porejestracyjne
badania skuteczności należy podjąć w przypadku
pojawienia się wątpliwości dotyczących konkretnych
aspektów skuteczności leku, na przykład w razie zmiany standardów postępowania w danej
chorobie lub udostępnienia nowych dowodów naukowych
podważających dotychczasową wiedzę.
Tego typu badań nie wykonuje się zatem zbyt często.
Komunikaty bezpieczeństwa to informacje
kierowane do lekarzy (zwykle lekarzy konkretnej
specjalności), a także – chociaż rzadko – do ogółu
społeczeństwa, gdy trzeba szybko poinformować o nowym, dotychczas nieznanym istotnym zagrożeniu
mającym związek z terapią. URPL publikuje
takie dokumenty na swojej stronie internetowej.
Są one ogólnodostępne pod adresem: www.urpl.gov.pl w zakładce „Komunikaty bezpieczeństwa”.
Materiały edukacyjne są przygotowywane
przez podmioty odpowiedzialne i zatwierdzane
przez URPL. W wielu przypadkach informują,
jak uniknąć lub ograniczyć ryzyko związane z terapią
poprzez wdrożenie odpowiednich działań
(np. przeprowadzenie właściwych badań diagnostycznych). W niektórych sytuacjach nie można
zwiększyć bezpieczeństwa, a jedynie poinformować o istniejącym ryzyku. Tak jest w przypadku
materiałów dotyczących szczepionki przeciwko
żółtej gorączce, w których opisano znane i potencjalnie
zagrażające życiu zagrożenia (choroba wiscerotropowa i choroba neurotropowa).
Każdy lek, a więc także szczepionka, może pozostać w obrocie dopóki korzyści z jego stosowania
przewyższają ryzyko. Obowiązek ciągłego nadzoru
spoczywa na instytucjach państwowych, takich
jak URPL, a także na firmach farmaceutycznych,
które przygotowują szereg dokumentów i analiz
dotyczących własnych produktów. Do podstawowych
materiałów, które przygotowuje się dla
każdego leku, należy plan zarządzania ryzykiem, a także dla wybranych produktów – okresowy
raport o bezpieczeństwie stosowania. Plan zarządzania
ryzykiem dotyczy przyszłości – z jednej
strony definiując ryzyko, a z drugiej proponując
kroki, jakie należy podjąć, aby uniknąć zagrożeń
lub je przynajmniej ograniczyć. Natomiast
okresowy raport o bezpieczeństwie stosowania
podsumowuje wiedzę o NDL zebranych w danym
przedziale czasu i analizuje, czy w świetle tych informacji
zmienia się ocena wartości leku, patrząc
na spodziewane korzyści z jego podania i możliwe
do przewidzenia ryzyko, jakie ze sobą niesie.
Działania podejmowane w ramach nadzoru
nad bezpieczeństwem mają od początku wymiar
globalny. Polska od początku zaangażowania się w monitorowanie działań niepożądanych działa w ramach programu opracowanego przez WHO.
Jednostka pracująca w URPL jest narodowym
ośrodkiem WHO.
Od czasu wejścia Polski do UE, URPL uczestniczy w wymianie informacji i ocenie danych zbieranych w poszczególnych krajach członkowskich.
Współpraca międzynarodowa, szybko postępująca
informatyzacja, stworzenie ogromnych baz danych, a także opracowane w ramach UE przepisy i procedury
pozwalają na szybką reakcję w przypadku
wykrycia nowego zagrożenia.
Podsumowanie
Szczepionki są produktami leczniczymi pochodzenia
biologicznego, podlegają wielu rygorystycznym
wymogom zarówno w zakresie produkcji, jak i kontroli
przez instytucje o charakterze rządowym lub
międzynarodowym. Istotnym etapem zapewniającym
wysoką jakość, bezpieczeństwo i skuteczność
szczepionek jest proces ich rejestracji. Pozwolenie
na dopuszczenie do obrotu jest przyznawane
po wnikliwej ocenie dokumentacji, która musi być
przygotowana zgodnie z wytycznymi europejskimi.
Znając pewne ograniczenia, jakim podlegają
badania kliniczne stanowiące podstawę do rejestracji
produktów leczniczych, konieczne jest ciągłe
monitorowanie danych dotyczących stosowania
szczepionek w codziennej praktyce klinicznej.
Pozwala to na bieżąco uzupełniać wiedzę i podejmować
odpowiednie kroki w przypadku wykrycia
zagrożeń.
Piśmiennictwo:
1. Ustawa z dnia 6 września 2001 roku Prawo farmaceutyczne. Dz. U. 2001, nr 126, poz. 13812. Ustawa z dnia 18 marca 2011 roku o Urzędzie Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych. Dz. U. 2011, nr 82, poz. 451
3. Dyrektywa 2001/83/WE Parlamentu Europejskiego i Rady z dnia 6 listopada 2001 roku w sprawie wspólnotowego kodeksu odnoszącego się do produktów leczniczych stosowanych u ludzi, Dz. U., L 311 z 28.11.2001, s. 67)
4. Rozporządzenie Ministra Zdrowia z dnia18 kwietnia 2014 roku w sprawie sposobu przedstawienia dokumentacji dołączonej do wniosku o dopuszczenie do obrotu produktu leczniczego, Dz. U. 2014, poz. 732
5. Rozporządzenie (WE) nr 726/2004 Parlamentu Europejskiego i Rady z dnia 31 marca 2004 r. ustanawiające wspólnotowe procedury wydawania pozwoleń dla produktów leczniczych stosowanych u ludzi i do celów weterynaryjnych i nadzoru nad nimi oraz ustanawiające Europejską Agencję Leków. Dz. U., L 136 z 30.04.2004, s. 1
6. European Medicines Agency. Guideline on clinical evaluation of new vaccines EMEA/CHMP/VWP/164653/2005
7. European Medicines Agency. Guideline on quality, non-clinical and clinical aspects of live recombinant viral vectored vaccines EMA/CHMP/VWP/141697/2009
8. European Medicines Agency. Guideline on adjuvants in vaccines for human use EMEA/CHMP/VEG/134716/2004
9. European Medicines Agency. Guideline on quality aspects included in the product information for vaccines for human use EMA/CHMP/BWP/133540/2017
10. European Pharmacopoeia/Farmakopea Polska
11. Rozporządzenie Ministra Zdrowia z dnia 21 grudnia 2010 roku w sprawie niepożądanych odczynów poszczepiennych oraz kryteriów ich rozpoznawania. Dz. U. 2010, nr 254, poz. 1711