Skróty: OMCL – Państwowe Laboratorium Kontroli Produktów Leczniczych, NIZP–PZH – Narodowy Instytut Zdrowia Publicznego–Państwowy Zakład Higieny, UE – Unia Europejska
Wprowadzenie
Kontrola jakości szczepionek obejmuje kontrolę
każdej serii szczepionki na każdym etapie wytwarzania,
przed zwolnieniem na rynek oraz w całym
okresie dostępności na rynku. Obowiązek kontroli
nakładają przepisy krajowe oraz Unii Europejskiej
(UE) zawarte m.in. w Ustawie Prawo Farmaceutyczne
oraz Dyrektywie 2001/83/EC Parlamentu
Europejskiego i Rady w sprawie wspólnotowego
kodeksu odnoszącego się do produktów leczniczych
stosowanych u ludzi, zmienionej przez Dyrektywę
2004/27/EC.
Niezależną od wytwórcy kontrolę przeprowadza
niezależne Państwowe Laboratorium Kontroli
Produktów Leczniczych (Official Medicines
Control Laboratory – OMCL). W Polsce
laboratorium odpowiedzialne za kontrolę jakości
szczepionek oraz zwolnienie każdej serii spełniającej
wymagania jakościowe na rynek jest wskazane w Rozporządzeniu Ministra Zdrowia z dnia
10 kwietnia 2013 roku w sprawie kontroli seryjnej
wstępnej produktów leczniczych oraz surowców
wykorzystywanych do sporządzania leków recepturowych
lub aptecznych, a także w Rozporządzeniu
Ministra Zdrowia z dnia 1 sierpnia 2016 roku w sprawie jednostek organizacyjnych, które prowadzą
badania jakościowe produktów leczniczych i produktów leczniczych weterynaryjnych, oraz
opłat pobieranych za te badania. Laboratorium
tym jest Zakład Badania Surowic i Szczepionek Narodowego Instytutu Zdrowia Publicznego–Państwowego Zakładu Higieny
(NIZP–PZH) w Warszawie i jest to jedyne laboratorium w Polsce, które posiada kompetencje w tym zakresie.
Kontrola jakości i bezpieczeństwa szczepionek przed wprowadzeniem do obrotu
Od rozpoczęcia badań nad opracowaniem nowej szczepionki do chwili jej wprowadzenia do obrotu upływa średnio 12–15 lat. W tym czasie podejmowane są niezbędne badania przedkliniczne oraz kliniczne. W ramach trwających 3–6 lat badań przedklinicznych dobierane są antygeny i technologia, a także wykonywane są badania dotyczące farmakodynamiki, skuteczności i bezpieczeństwa in vitro oraz in vivo. Jeśli uzyskane wyniki są zgodne z oczekiwaniami i spełniają określone wymogi, rozpoczyna się I faza badań klinicznych, która trwa zwykle 12–18 miesięcy. Podczas tej fazy badań szczepionkę podaje się 20–50 zdrowym ochotnikom i ocenia się jej bezpieczeństwo, dobiera odpowiednią dawkę oraz identyfikuje się ewentualne skutki uboczne i niepożądane odczyny poszczepienne. Wyniki I fazy dostarczają także wstępnych danych dotyczących dawkowania i czasu między podaniem szczepionki a uzyskaniem optymalnej odpowiedzi immunologicznej. II faza badań klinicznych obejmuje już większą liczbę ochotników, tj. 100–300 zdrowych osób, i trwa co najmniej 2 lata. W ramach tej fazy kontynuuje się ocenę bezpieczeństwa i immunogenności szczepionki oraz opracowuje się schemat dawkowania. Faza III badań klinicznych trwa zwykle 3–5 lat i obejmuje dużą, liczącą 3000–50 000 grupę zdrowych ochotników. Jej celem jest dalsze monitorowanie bezpieczeństwa, immunogenności i skuteczności szczepionki, a także ocena jej jednoczesnego stosowania z innymi szczepionkami. Przejście do każdej kolejnej fazy badań jest uwarunkowane uzyskaniem wyników zgodnych oczekiwaniami i wymogami w zakresie m.in. skuteczności, jakości i bezpieczeństwa. Uproszczony schemat przebiegu prac nad opracowaniem szczepionki przedstawiono na rycinie 1.
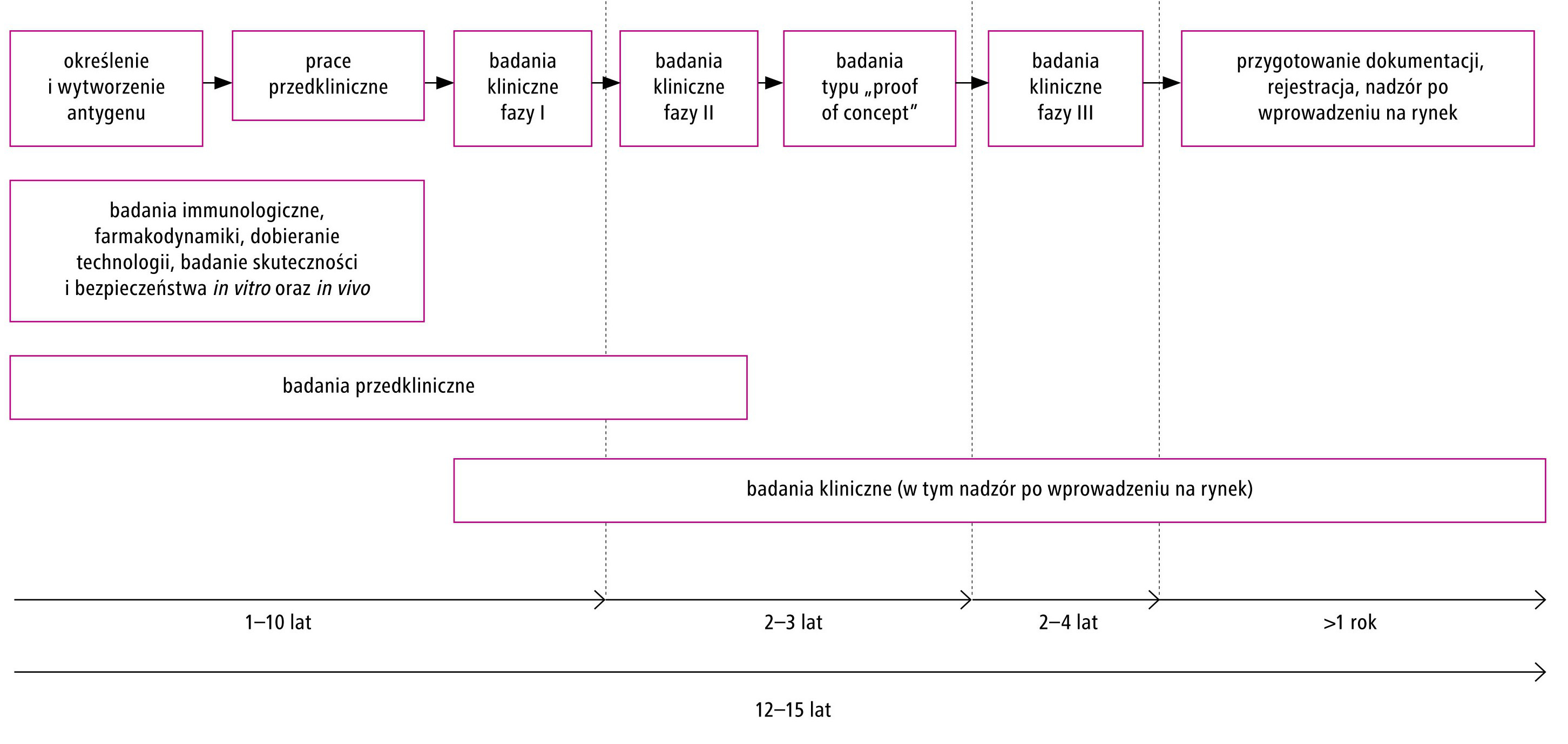
Ryc. 1. Uproszczony schemat przebiegu prac nad opracowaniem szczepionki.
Wprowadzenie nowej szczepionki do obrotu
Szczepionkę, która przeszła pozytywnie wszystkie
etapy badań (p. ryc. 1.), można zgłosić do rejestracji i ubiegania się o pozwolenie na dopuszczenie
do obrotu. Rejestracja i dopuszczenie do obrotu
nowego produktu leczniczego (w tym przypadku
szczepionki) może się odbywać w ramach procedury
scentralizowanej, narodowej, procedury
zdecentralizowanej lub procedury wzajemnego
uznania. Procedurę scentralizowaną przeprowadza
Europejska Agencja Leków (European Medicines
Agency – EMA) i wiąże się ona z rejestracją i dopuszczeniem do obrotu szczepionki na terenie
całej UE. W Polsce procedura narodowa przeprowadzana
jest przez Urząd Rejestracji Produktów
Leczniczych, Wyrobów Medycznych i Produktów
Biobójczych (URPL). W procedurze wzajemnego
uznania produkt najpierw jest rejestrowany
na poziomie narodowym, a następnie rejestracja
jest rozszerzana na inne kraje UE (p. Med. Prakt. Szczepienia 3/2019, s. 97 – przyp. red.).
W trakcie procedury rejestracji i dopuszczenia
do obrotu nowej szczepionki eksperci analizują
wiele aspektów. Oceniany jest stosunek korzyści
do ryzyka (risk-benefit ratio) związanego ze stosowaniem
nowego produktu oraz analizowane są
wszystkie dane zebrane podczas rozwoju produktu i badań klinicznych. Ocenie podlega też jakość,
bezpieczeństwo i skuteczność nowej szczepionki.
Weryfikowana jest zgodność z dobrą praktyką wytwarzania
(Good Manufacturing Practice – GMP),
dobrą praktyką badań klinicznych (Good Clinical
Practice – GCP) oraz dobrą praktyką laboratoryjną
(Good Laboratory Practice – GLP). Oceniana
jest również potrzeba populacji dla wprowadzenia
nowej szczepionki.
Kontrola jakości i bezpieczeństwa szczepionek przez wytwórcę
Wytwórca szczepionki na obowiązek prowadzić kontrolę jakości na każdym etapie jej wytwarzania oraz każdego materiału wyjściowego wykorzystywanego w procesie produkcji szczepionki. Kontrolowane są m.in. wszystkie pożywki hodowlane, linie komórkowe, odczynniki chemiczne oraz szczepy bakterii i wirusów używane do produkcji. Kontrola na każdym etapie produkcji obejmuje m.in. kontrolę substancji aktywnych, zawartości poszczególnych składników szczepionki oraz czystości. Na koniec wytwórca przeprowadza kontrolę produktu końcowego, czyli gotowej, ostatecznej postaci szczepionki. Jeśli wszystkie wyniki badań są zgodne ze specyfikacją, wytwórca lub podmiot odpowiedzialny zgłasza daną serię szczepionki do kontroli seryjnej wstępnej prowadzonej przez OMCL. Sam wytwórca jest również kontrolowany w zakresie GMP przez odpowiednie organy państwowe. W Polsce kontrolę taką przeprowadza Główny Inspektorat Farmaceutyczny (GIF).
Kontrola jakości szczepionek przez niezależne Państwowe Laboratorium Kontroli Produktów Leczniczych
Seria szczepionki, która przeszła pozytywnie wszystkie badania jakości u wytwórcy, jest przekazywana do niezależnego OMCL do kontroli seryjnej wstępnej (nazwa „kontrola seryjna wstępna” odnosi się do „wstąpienia” produktu na rynek). W ramach tej kontroli przeprowadza się szereg badań laboratoryjnych zgodnie z wytycznymi dla danego rodzaju szczepionki, które mogą obejmować badania fizykochemiczne, biochemiczne, biologiczne, serologiczne, mikrobiologiczne i molekularne. Weryfikowana jest również dokumentacja wytwórcy. Minimalny zakres badań, jaki OMCL musi wykonać, aby dana seria szczepionki trafiła na rynek, określają wytyczne Europejskiego Dyrektoriatu ds. Jakości Leków (European Directiorate for the Quality of Medicines – EDQM) osobno dla każdego rodzaju szczepionki. Badania te są dobierane na podstawie składu szczepionki, procedury jej wytwarzania oraz oceny ryzyka uwzględniającej m.in. newralgiczne etapy produkcji, które mogą mieć wpływ na jakość szczepionki, a tym samym jej bezpieczeństwo i skuteczność. W tabeli wymieniono przykłady badań wykonywanych w Zakładzie Badania Surowic i Szczepionek NIZP–PZH dla szczepionki przeciwko błonicy, tężcowi i krztuścowi (DTP), gruźlicy (BCG) oraz wirusowemu zapaleniu wątroby typu B.
| Tabela. Zestaw badań jakościowych wykonywanych rutynowo w NIZP–PZH na przykładzie szczepionki DTP, BCG oraz przeciwko WZW typu B | ||
|---|---|---|
| DTP | BCG | WZW typu B |
| wygląd zanieczyszczenia cząstkami widocznymi okiem nieuzbrojonym objętość płynu uzyskiwana z pojemnika aktywność składnika błoniczego, krztuścowego, tężcowego toksyczność składnika krztuścowego pH zawartość glinu zawartość wolnego formaldehydu |
wygląd wygląd po rekonstytucji tożsamość liczba żywych cząstek BCG termostabilność |
wygląd zanieczyszczenia cząstkami widocznymi okiem nieuzbrojonym objętość płynu uzyskiwana z pojemnika zawartość glinu zawartość wolnego formaldehydu aktywność szczepionki (moc) |
| BCG – szczepionka przeciwko gruźlicy, DTP – szczepionka przeciwko błonicy, tężcowi i krztuścowi, NIZP–PZH – Narodowy Instytut Zdrowia Publicznego–Państwowy Zakład Higieny, WZW – wirusowe zapalenie wątroby | ||
Jeżeli wyniki badania przeprowadzonego w OMCL są zgodne ze specyfikacją (jeden z dokumentów
wymaganych przy rejestracji produktu
udostępniany OMCL przez URPL), seria szczepionki
uzyskuje pozwolenie na zwolnienie na rynek.
Natomiast szczepionka, której chociaż jeden
parametr specyfikacji jest niespełniony, nie uzyskuje
pozwolenia na zwolnienie, o czym powiadamiany
jest GIF oraz wszystkie laboratoria OMCL w Europie, aby zapobiec ewentualnemu pojawieniu
się tej serii szczepionki w innym kraju UE. W związku z przyjętą zasadą wzajemnego uznawania wyników badań przez laboratoria OMCL w Europie, serii szczepionki, która nie przeszła
pozytywnie badań w jednym z OMCL, nie można
zgłosić do badania w innym OMCL (np. w innym
kraju). Cała seria szczepionki, która nie uzyskała
pozwolenia na zwolnienie na rynek, zostaje poddana
utylizacji.
Serie szczepionki zwolnione na rynek nadal
podlegają kontroli na terenie całej UE. W Polsce
kontrolę taką przeprowadza OMCL na zlecenie
GIF i polega ona na wyrywkowej kontroli szczepionek
dostępnych w hurtowniach, aptekach lub
punktach szczepień. GIF może również zlecić kontrolę
po zgłoszeniu przez lekarza lub pielęgniarkę
wykonującą szczepienie podejrzenia niespełnienia
wymagań (np. wygląd szczepionki nietypowy lub
niezgodny z opisem w ulotce). W obydwu przypadkach
badania rozpoczyna się niezwłocznie po dostarczeniu
odpowiedniej ilości produktu do laboratorium
OMCL i wykonuje pełny panel badań,
taki jak podczas kontroli seryjnej wstępnej przed
zwolnieniem serii szczepionki na rynek. W przypadku
podejrzenia niespełnienia wymagań GIF
może podjąć decyzję o wstrzymaniu danej serii
szczepionki w obrocie do czasu uzyskania wyników
badań. Jeśli wyniki badań potwierdzą wadliwość
danej serii szczepionki, jest ona wycofywana z rynku i przekazywana do utylizacji. Uproszczony
schemat ciągłej kontroli nad każdą serią szczepionki
przed zwolnieniem na rynek oraz po zwolnieniu
na rynek przedstawiono na rycinie 2.
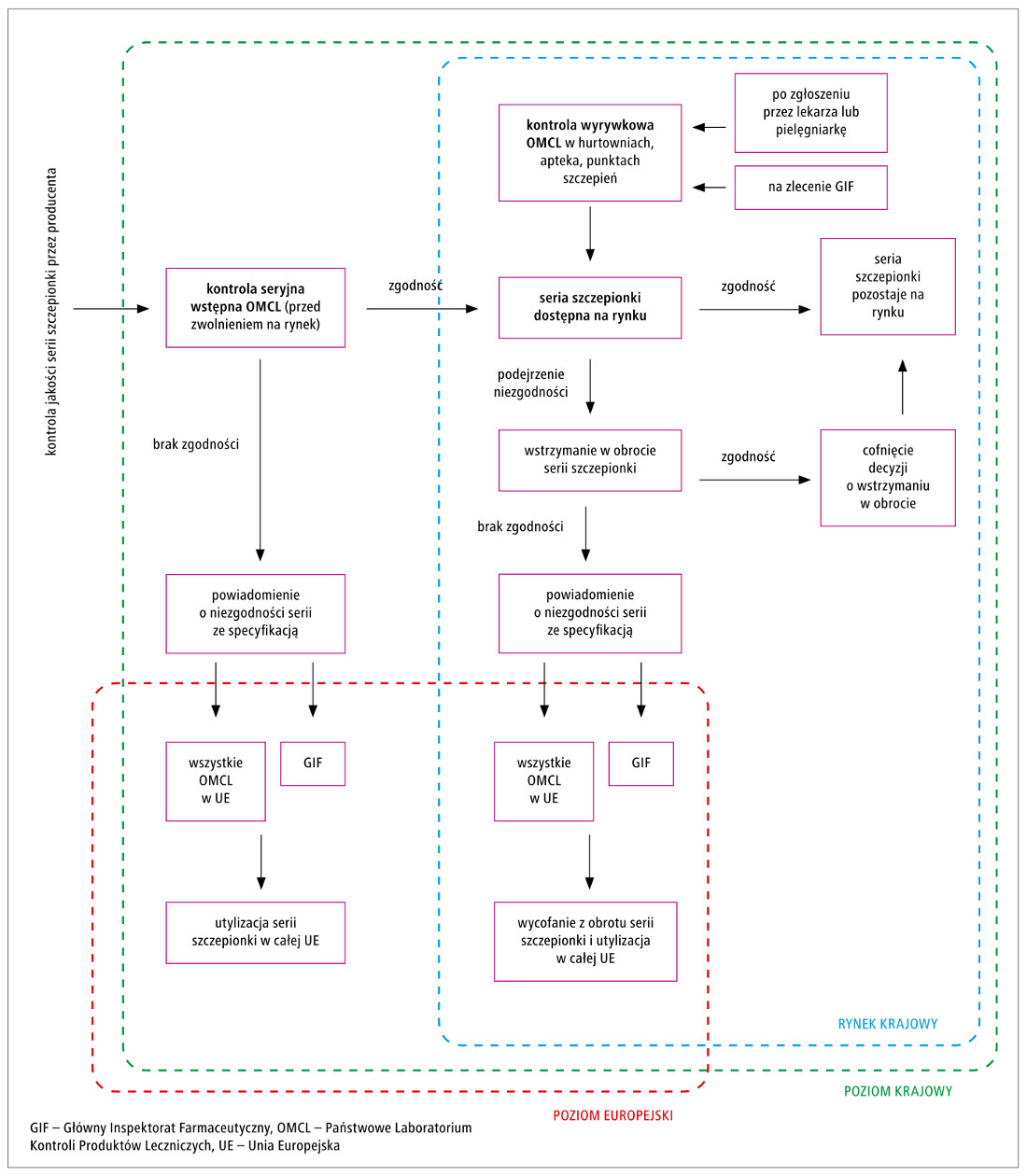
Ryc. 2. Uproszczony schemat kontroli jakości i bezpieczeństwa szczepionek przez niezależne OMCL w Polsce.
Wymagania dla niezależnego Państwowego Laboratorium Kontroli Produktów Leczniczych
Aby pełnić funkcję niezależnego OMCL, należy spełniać szereg bardzo restrykcyjnych wymagań. Obejmują one m.in. system jakości ISO 17 025, krajową akredytację jednostki oraz certyfikację na poziomie europejskim. System jakości ISO 17 025 określa szereg wymagań w różnych obszarach, m.in. w zakresie poufności i bezstronności, posiadanych zasobów (personelu, pomieszczeń i warunków środowiska, wyposażenia, spójności pomiarowej, wyrobów i usług dostarczanych z zewnątrz), procesu (np. postępowania z obiektami do badań, zapisów technicznych, oceny niepewności pomiarów, potwierdzania ważności wyników, raportowania wyników, nadzorowania danych i zarządzania informacją), a także wymagań dotyczących systemu zarządzania (np. nadzór nad dokumentami i zapisami, działania odnoszące się do ryzyka i szans, doskonalenie, działania korygujące, audyty wewnętrzne). Uzyskanie akredytacji ISO 17 025 przez laboratorium jest potwierdzeniem, że wykonywane przez to laboratorium badania są zgodne z systemem jakości, a otrzymywane wyniki są miarodajne, charakteryzują się rzetelnością i dokładnością. W Polsce niezależne państwowe laboratorium przeprowadzające kontrolę szczepionek, którym jest Zakład Badania Surowic i Szczepionek NIZP–PZH, posiada akredytację Polskiego Centrum Akredytacji (PCA) oraz certyfikację Europejskiego Dyrektoriatu ds. Jakości Leków (EDQM). Aby uzyskać i utrzymać atestację oraz certyfikację laboratorium, musi m.in. posługiwać się w badaniach zwalidowanymi metodami, a badania może wykonywać wyłącznie wykwalifikowany personel, który przechodzi regularne szkolenia. Laboratorium musi brać regularny udział w ocenie biegłości w zakresie stosowanych metod badawczych i przechodzić je z pozytywnym wynikiem. Całe wyposażenie wykorzystywane do badań musi być regularnie wzorcowane, sprawdzane i kwalifikowane do użytku. Laboratorium musi także posiadać i stosować odpowiednie procedury postępowania w przypadku uzyskania wyników niezgodnych ze specyfikacją danej szczepionki. Ma obowiązek także prowadzić analizę trendu danych uzyskiwanych w badaniach. Ponadto OMCL musi wykazać całkowitą niezależność i brak konfliktu interesów w zakresie swojej działalności, na przykład pracownicy nie mogą pozostawać w związkach rodzinnych ani służbowych z firmami farmaceutycznymi i ich pracownikami. Należy podkreślić, że OMCL jest regularnie kontrolowane przez różne podmioty państwowe i międzynarodowe, na przykład GIF, EDQM oraz PCA.
Podsumowanie
Z całą pewnością można stwierdzić, że szczepionki są najlepiej kontrolowanym i tym samym najbezpieczniejszym produktem leczniczym nie tylko w Polsce, ale i całej Europie. Jest to jeden z nielicznych produktów leczniczych, którego każda seria – przed zwolnieniem na rynek – podlega kontroli przez niezależne od wytwórcy państwowe laboratorium, a stale monitorowana jakość prowadzonej kontroli jest na bardzo wysokim poziomie.
Piśmiennictwo:
1. Ustawa z dnia 6 września 2001 r. Prawo farmaceutyczne. Dz. U. 2001 nr 126, poz. 13812. Dyrektywa 2001/83/WE Parlamentu Europejskiego I Rady z dnia 6 listopada 2001 r. w sprawie wspólnotowego kodeksu odnoszącego się do produktów leczniczych stosowanych u ludzi. Dz. U. L 311 z 28.11.2001, s. 67
3. Dyrektywa 2004/27/WE Parlamentu Europejskiego i Rady z dnia 31 marca 2004 r. zmieniająca dyrektywę 2001/83/WE w sprawie wspólnotowego kodeksu odnoszącego się do produktów leczniczych stosowanych u ludzi
4. Rozporządzenie Ministra Zdrowia z dnia 10 kwietnia 2013 r. w sprawie kontroli seryjnej wstępnej produktów leczniczych oraz surowców wykorzystywanych do sporządzania leków recepturowych lub aptecznych. Dz. U. 2013, poz. 491
5. Rozporządzenie Ministra Zdrowia z dnia 1 sierpnia 2016 r. w sprawie jednostek organizacyjnych, które prowadzą badania jakościowe produktów leczniczych i produktów leczniczych weterynaryjnych, oraz opłat pobieranych za te badania. Dz. U. 2016, poz. 1179
6. European Medicines Agency. Human medicines: regulatory information. www.ema.europa.eu/en
7. Vaccines Europe. How are vaccines developed? www.vaccineseurope.eu
8. European Directorate for the Quality of Medicines and Health Care (EDQM). Batch Release for Human Biologicals: Vaccines, blood and plasma derivatives. www.edqm.eu
9. World Health Organization (WHO). Immunization standards. Vaccine regulation. www.who.int
10. Vaccines Europe. EU regulatory framework for vaccines. www.vaccineseurope.eu
11. World Health Organization (WHO). Guidelines for Independent Lot Release of Vaccines by Regulatory Authorities. WHO 2010, Geneva, Switzerland